
肿瘤纯度和倍性评估工具Sequenza的安装和使用方法
肿瘤样本中癌细胞总是混合一定未知比例的正常细胞,我们称肿瘤样本中癌细胞所占的比例为肿瘤纯度(Tumor purity),称由染色体结构和数目异常导致的肿瘤样本中癌细胞的真正含量为倍性(Tumor ploidy)。估计肿瘤的纯度和倍性有利于癌症基因组进化和肿瘤内的异质性研究。
Sequenza是一个使用配对的肿瘤/正常样本DNA测序数据来估计肿瘤样本纯度和倍性的软件,同时还检测肿瘤样本拷贝数变异。本期小编详细给大家介绍该软件的安装及使用。
1 软件安装
首先是软件安装,该软件依赖python包sequenza-utils和R包sequenza,分别安装这两个包,有很多小伙伴在安装软件的过程中总会遇到各种版本问题,建议使用conda虚拟环境安装,首先在虚拟环境中安装python和R:
# 创建虚拟环境Sequenza,并安装python
conda create -n Sequenza python=3.8
# 为虚拟环境安装R
conda install -n Sequenza r=3.6.3
# 进入虚拟环境Sequenza
conda activate Sequenza
然后就是在虚拟环境中进行python和R包安装,跟在linux环境下安装一样。
1.1.sequenza-utils安装(python包)
sequenza-utils包安装比较容易,以下有两种安装方式:
官方说明:https://sequenza-utils.readthedocs.io/en/latest/
# 安装方法1(推荐)
pip install sequenza-utils
# 安装方法2
git clone https://bitbucket.org/sequenza_tools/sequenza-utils
cd sequenza-utils
python setup.py test
python setup.py install
# 安装成功后,查看帮助
sequenza-utils -h
1.2.sequenza安装(R包)
# 安装sequenza包前,需要先安装copynumber包
install.packages("BiocManager")
library(BiocManager)
BiocManager::install("copynumber")
# 或者使用conda安装(推荐)
conda install -c bioconda bioconductor-copynumber
# 安装sequenza包
install.packages("sequenza")
# 安装成功后,查看帮助
library(sequenza)
library(help="sequenza")
2 使用方法
Python和R包安装成功后,就可以进行肿瘤纯度和倍性评估,我们使用处理后的肿瘤配对样本BAM文件、参考基因组ref文件作为输入,BAM文件通过基因组比对流程得到(如GATK标准流程),参考基因组文件可以通过UCSC下载(hg19/hg38)。
2.1. 基于python的预处理sequenza-utils
# 制作参考基因组GC标准化文件,通过-w参数分割基因组文件,设置越小检测敏感性越高。
sequenza-utils gc_wiggle -f $ref -w 50 -o - | gzip > hg19.gc50Base.txt.gz
# 根据肿瘤样本和对照样本BAM文件统计GC标准文件每个碱基的深度和等位碱基频率等。
sequenza-utils bam2seqz \
-gc $out/hg19.gc50Base.txt.gz \
-F $ref \
-n $normalbam \
-t $tumorbam | gzip >$out/$tumor/${tumor}.seqz.gz \
# 提取测序深度,确定正常标本中的纯合和杂合位置,并从肿瘤标本中计算出变异等位基因和等位基因频率。减小seqz文件的大小,提高模型运行效率
sequenza-utils seqz_binning -w 50 \
-s $out/$tumor/${tumor}.seqz.gz | gzip >$out/$tumor/${tumor}_small.seqz.gz
2.2. 模型拟合及可视化工具sequenza
# 输入文件(${tumor}_small.seqz.gz)预处理
(1)需要删除chrM或非常规染色体数据,否则会报错xlim有非限制值;
(2)为了提高评估准确性,可以对数据进行筛选,比如筛选DP > 10。
这个处理步骤自行使用python或R编程,然后再压缩,名字还是一样。
# 导入预处理数据,并对肿瘤进行GC含量归一化与正常深度之比,并使用“copynumber”软件包进行等位基因特异性分割。
test <- sequenza.extract(“${tumor}_small.seqz.gz”, verbose = FALSE)
# 推断细胞性和倍性参数以及拷贝数分布图,使用后验概率空间的局部最大值来提供替代解决方案
CP <- sequenza.fit(test)
#计算纯度和倍性,很耗时间
# 返回估计结果以及替代解决方案以及沿基因组和单个染色体的数据和模型的可视化
sequenza.results(sequenza.extract = test,
cp.table = CP,
sample.id = sample,
out.dir=sample)
3 结果说明
所有的结果文件说明如下:

结果有很多,但是纯度和倍性的相关结果其实就3个文件,如下:

我们依次打开这三个文件,一起来看看结果长啥样:
(1)result_alternative_solutions.txt
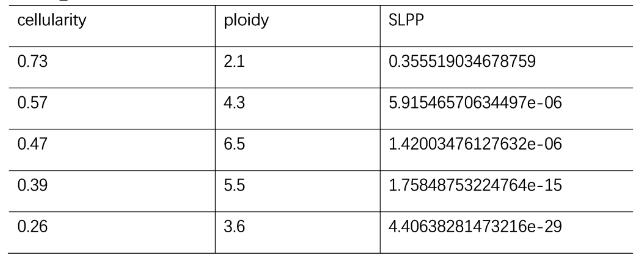
表1 肿瘤样本纯度和倍性评估结果
说明:Cellularity:肿瘤样本纯度 Ploidy:肿瘤样本倍性 SLPP:对数后验概率
(2)result_CP_contours.pdf
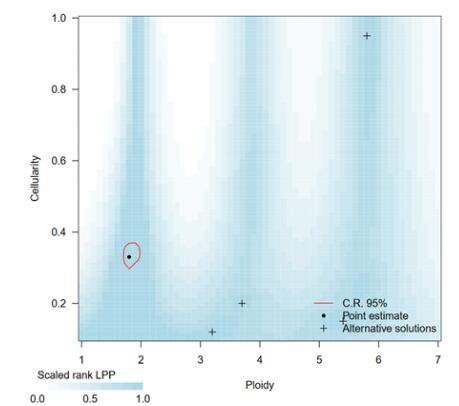
图1 肿瘤样本纯度与模型结果
说明:横坐标为倍性值,纵坐标为纯度值,背景蓝色表示最有可能的分布,白色表示最不可能的分布,其中红色圈点为最优值,即SLPP值最大,其它为次优解(图中“+”)
(3)result_model_fit.pdf
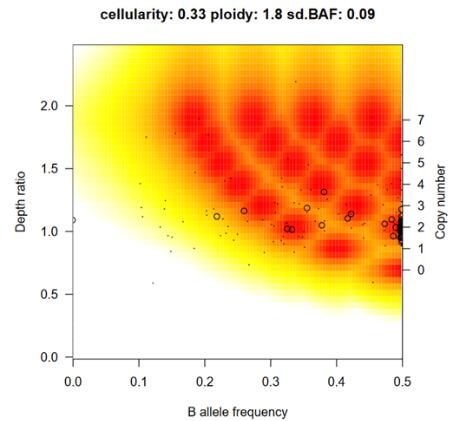
图2 肿瘤样本倍性模型评估结果
说明:横坐标为B allele frequency,纵坐标左侧Depth ratio代表每个基因组片段肿瘤样本与正常样本测序深度比(低比值下估算的肿瘤样本拷贝数不可信),纵坐标右侧copy number代表模型估算的拷贝数(黑色圆圈和点),背景颜色越红表示越可信。
好了,以上就是使用基因组数据评估肿瘤样本纯度和倍性的过程。更多实用生信分析方法,小编将持续更新。



