
DeepSearch――基于深度学习的高灵敏串联质谱数据搜库分析策略
在基于质谱(MS)的蛋白质组学中,肽段鉴定是关键挑战。传统数据库搜索方法依赖启发式评分函数,存在对某些肽段组成的偏差,需引入统计估计提高鉴定率。深度学习虽提升了肽段从头测序的准确性,但现有方法在处理不同蛋白质组成数据集及鉴定可变翻译后修饰(PTM)方面仍存在不足。
为应对以上问题,Yonghan Yu和李明教授(Bioinformatics Solution Inc.创始人、加拿大皇家学会院士)在Nature Machine Intelligence(IF 18.8)发表了题为“Towards highly sensitive deep learning-based end-to-end database search for tandem mass spectrometry”的最新研究成果,提出了一种新的串联质谱数据库搜索方法—DeepSearch。在对比学习框架下,DeepSearch采用了改进的基于Transformer 的编解码器架构。与传统的离子与离子匹配方法不同,DeepSearch 采用数据驱动的方法对肽段-谱图匹配进行评分,显著降低了评分偏差,并且支持零样本变量的可变翻译后修饰(PTM)鉴定。DeepSearch在各种数据集上均表现出较高的准确性和稳定性,包括不同物种的数据集以及富含PTM的数据集等。DeepSearch 为串联质谱的数据库搜索方法提供了新的思路。
为应对以上问题,Yonghan Yu和李明教授(Bioinformatics Solution Inc.创始人、加拿大皇家学会院士)在Nature Machine Intelligence(IF 18.8)发表了题为“Towards highly sensitive deep learning-based end-to-end database search for tandem mass spectrometry”的最新研究成果,提出了一种新的串联质谱数据库搜索方法—DeepSearch。在对比学习框架下,DeepSearch采用了改进的基于Transformer 的编解码器架构。与传统的离子与离子匹配方法不同,DeepSearch 采用数据驱动的方法对肽段-谱图匹配进行评分,显著降低了评分偏差,并且支持零样本变量的可变翻译后修饰(PTM)鉴定。DeepSearch在各种数据集上均表现出较高的准确性和稳定性,包括不同物种的数据集以及富含PTM的数据集等。DeepSearch 为串联质谱的数据库搜索方法提供了新的思路。
DeepSearch方法
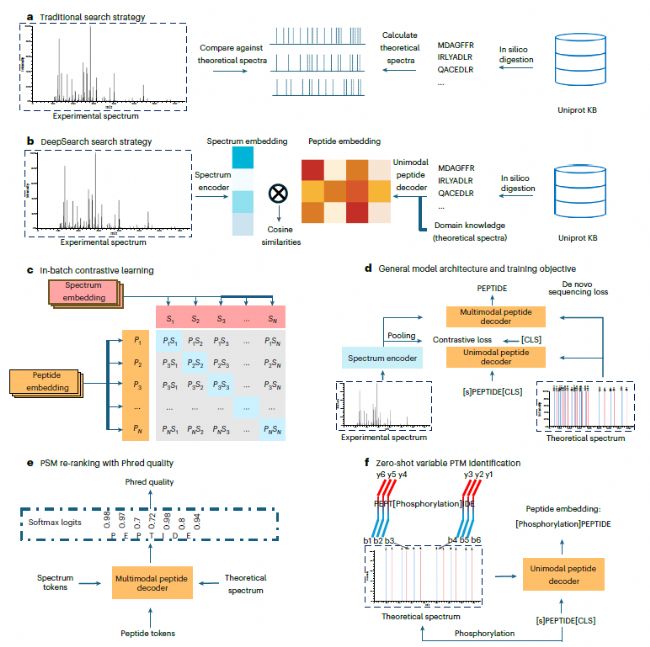
图1 DeepSearch搜索模型
传统的谱图搜索策略一般是将标准参考序列通过计算机模拟酶切(in-silico digestion)后,生成对应的理论谱,然后与实际采集的谱图进行匹配。因此,对于复杂谱图来说,就存在一些局限性。
DeepSearch采用改进的基于Transformer的编解码器架构,从蛋白质数据库的计算机理论酶解开始,DeepSearch将酶解的肽和实验MS/ MS谱图编码到嵌入中。DeepSearch不依赖于离子间匹配的启发式评分函数,而是使用相应嵌入之间的余弦相似性来对PSM进行评分,这可以通过单个矩阵乘法有效地计算出来。
为了解决在 PSM 中注释密切相关的负对的挑战,并减少注释中采用的搜索引擎的偏差,DeepSearch采用了批量内对比学习框架 。在训练过程中,DeepSearch 随机对一批锚定肽段质量的PSM 进行采样(正对),并将肽段-谱对(不包括采样的 PSM)用作负对(图 1c),通过对比学习,使正对之间的余弦相似度更高,而负对之间的余弦相似度更低。并且,DeepSearch 通过Phred分数对PSMs进行重排序,确保最终的匹配更加准确。在理论谱图中引入修饰质量偏移(mass shift),生成包含修饰信息的肽段嵌入。通过对比学习,DeepSearch可以直接对具有不同修饰的肽段进行鉴定。
实验结果
1. PSM 评分偏差较小
拟南芥数据集的测试结果显示,与 MSFragger、MS- GF+和 MaxQuant 比较,DeepSearch 的评分不受肽段长度影响,对缺失片段较多的短肽段评分较低,且在不同缺失片段数量下长肽段分数分布无显著差异。在 1% FDR 控制下,其报告的 PSM 数量与其他引擎相比具有优势,不受统计模型影响(图2a)。此外,目标序列匹配分数(蓝色)在所有肽段长度范围内分布均匀,表明DeepSearch的评分机制对肽段长度变化的稳定性。Decoy匹配(红色)较低且分布较窄,说明decoy匹配分数的波动较小,质控良好。
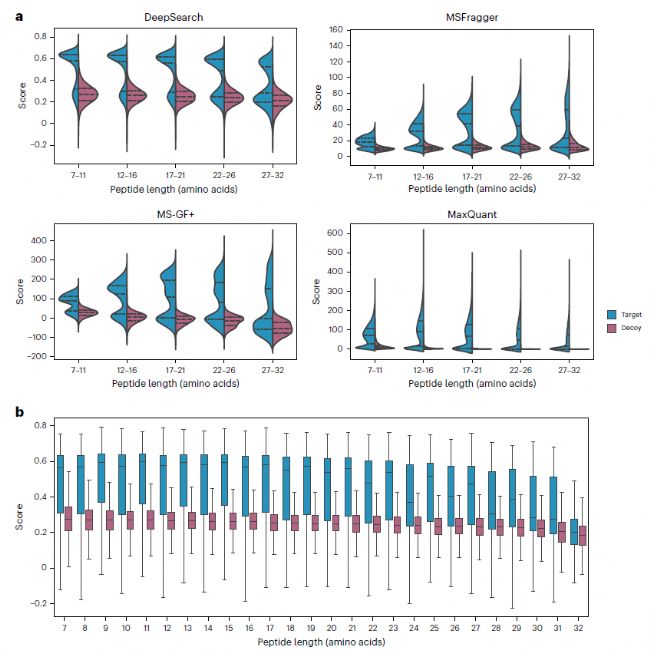 图2 不同搜索引擎对拟南芥数据集中不同长度肽段的鉴定
图2 不同搜索引擎对拟南芥数据集中不同长度肽段的鉴定2. 肽段鉴定准确且稳健
图3(a–d)分别展示了拟南芥(A. thaliana)、HEK293细胞、秀丽隐杆线虫(C. elegans)和大肠杆菌(E. coli)数据集在1%假阳性率(FDR)下的PSM数量。结果显示DeepSearch在不依赖统计模型的情况下,仍能维持较高的PSM鉴定数量,说明对于统计估计的依赖性已显著降低。
 图3 不同物种数据集通过FDR 1%质控的PSM数量
图3 不同物种数据集通过FDR 1%质控的PSM数量3. 零样本可变 PTM 分析
传统搜库方法通常需要提前对特定翻译后修饰(如磷酸化)的数据进行训练,限制了未知修饰的分析与发现。而DeepSearch借助深度学习,结合谱图与肽段序列之间的普遍规律,可以实现零样本的翻译后修饰训练。从图4 HeLa 磷酸化富集数据集的测试结果看,DeepSearch在零样本条件下,PTM分析的表现良好。图4a分别表示对于非修饰肽段、单位点修饰肽段、双位点修饰肽段的评分分布,可以看出随着修饰数量增加,目标肽段的匹配(蓝色)评分分布变宽,decoy匹配的得分分布变化较小,说明虽然修饰的复杂性对target匹配影响较大,但仍能保持较好的decoy质控。与MSFragger和MS-GF+相比,DeepSearch的准确性较高,但修饰肽的鉴定数量略少一些(图4b-d),有待进一步优化。
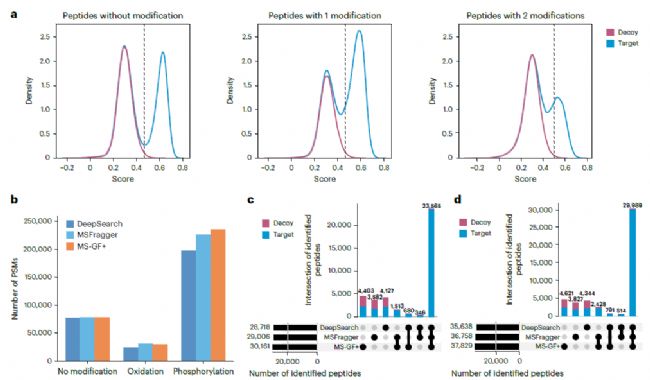
图4 Hela磷酸化富集数据集的零样本PTM表征
结论与展望
DeepSearch 是首个基于深度学习的端到端的串联质谱数据库搜索引擎,评分偏差小、准确性和稳健性高,能实现零样本PTM分析,标志着AI技术在蛋白质组学领域的重大应用突破。未来,DeepSearch有望作为独立引擎或重新评分模块,整合到现有蛋白质组学分析流程中,推动蛋白质组学尤其是复杂修饰组学的快速发展。
文献原文
Yu, Y., Li, M. Towards highly sensitive deep learning-based end-to-end database search for tandem mass spectrometry. Nat Mach Intell (2025). https://doi.org/10.1038/s42256-024-00960-1
作为生物信息学的领军企业,BSI专注于蛋白质组学和生物药领域,通过机器学习和先进算法提供世界领先的质谱数据分析软件和蛋白质组学服务解决方案,以推进生物学研究和药物发现。我们通过基于AI的计算方案,为您提供对蛋白质组学、基因组学和医学的卓越洞见。旗下著名的PEAKS®️系列软件在全世界拥有数千家学术和工业用户,包括:PEAKS®️ Studio,PEAKS®️ Online,PEAKS®️ GlycanFinder, PEAKS®️ AB,DeepImmu®️ 免疫肽组发现服务和抗体综合表征服务等。
标签:
蛋白质组学;串联质谱;搜库



