
利用深度学习框架实现肽段从头测序FDR和准确性评估详解
多肽从头测序是基于质谱的蛋白质组学的基本研究领域,对于鉴定不在任何数据库中的新型肽或蛋白质至关重要(例如突变的新抗原或抗体可变区)。在过去的三十年中,出现了数十种算法。Bittremieux等人最近的一篇综述论文对多肽从头测序的深度学习方法进行了全面介绍[1],可点击跳转至本期推送查看详细解读(综述文章 | 多肽从头测序的深度学习方法)。但现有的众多算法对于de novo结果的评估均存在较大的局限性,如难以生成合适的decoy来估计FDR、现有的准确性指标定义和计算方法存在偏差、深度学习模型可能存在过拟合和记忆问题等。

因此,Bioinformatics Solutions Inc.的算法团队开发了NovoBoard算法框架,这是一个用于评估多肽从头测序方法性能的综合框架,相应文章“NovoBoard: a comprehensive framework for evaluating the false discovery rate and accuracy of de novo peptide sequencing”已上线MCP。该框架涵盖不同的基准数据集(包括胰酶、非酶切、免疫肽组学和不同物种)和一套标准的准确性指标,用于评估从头测序结果的碎片离子、氨基酸和多肽序列。更重要的是,NovoBoard通过全新的生成decoy spectrum的方法计算从头测序的FDR,为评估从头测序报告的新多肽的可靠性评估提供有价值的参考信息。该框架已整合入PEAKS®️ 12软件中,欢迎通过文末的联系方式与我们咨询与交流。
01从头测序的FDR计算
在常规搜库方法中,诱饵肽(decoy peptides)可以通过随机打乱或反转参考蛋白质的序列来生成。但对于从头测序的方法来讲,在没有参考序列的情况下显然这种方法并不适合。因此,NovoBoard通过诱饵谱(decoy spectrum)而不是decoy peptide的方法来计算从头测序的FDR。
给定一个质谱数据集,从target spectrum中移除一些峰,然后添加相同数量的噪音峰以生成对应的decoy spectrum(峰包含m/z和intensity两个指标)。该过程确保每个decoy spectrum的峰总数与其target spectrum相同,并且噪音峰是从与target spectrum相同的分布中随机抽取的(图1a-c)。另一个关键问题是,应该移除多少以及选择哪些峰去除。我们尝试了去除10%到90%不等的峰,另外对比了两种选择去除峰的方法。第一种方法是随机选择,不考虑峰的m/z或intensity。第二种方法是首先计算要去除的峰数,模拟对应肽段质量下理论上会产生的b/y离子数量,然后根据峰强度选择。结果显示基于intensity和肽质量去峰的方法比随机选择方法的从头测序准确性更高。生成decoy spectrums后,对所有target和decoy spectrums进行多肽从头测序,并比较它们的得分分布以估计 FDR(图1 d)。但是,由于每张MS/MS都会产生来自target各decoy的两个de novo PSM,因此保留得分较高的PSM也是一种可能考虑的方法(图1 e)。我们对比了基于图1d的总体FDR和图1e的选择性PSM FDR,结果显示后者的FDR更严格。
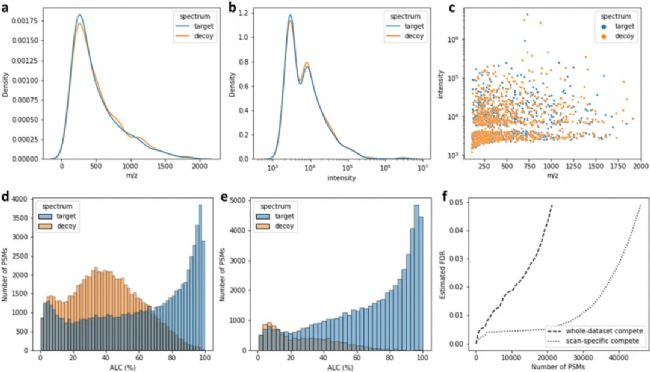
给定一个质谱数据集,从target spectrum中移除一些峰,然后添加相同数量的噪音峰以生成对应的decoy spectrum(峰包含m/z和intensity两个指标)。该过程确保每个decoy spectrum的峰总数与其target spectrum相同,并且噪音峰是从与target spectrum相同的分布中随机抽取的(图1a-c)。另一个关键问题是,应该移除多少以及选择哪些峰去除。我们尝试了去除10%到90%不等的峰,另外对比了两种选择去除峰的方法。第一种方法是随机选择,不考虑峰的m/z或intensity。第二种方法是首先计算要去除的峰数,模拟对应肽段质量下理论上会产生的b/y离子数量,然后根据峰强度选择。结果显示基于intensity和肽质量去峰的方法比随机选择方法的从头测序准确性更高。生成decoy spectrums后,对所有target和decoy spectrums进行多肽从头测序,并比较它们的得分分布以估计 FDR(图1 d)。但是,由于每张MS/MS都会产生来自target各decoy的两个de novo PSM,因此保留得分较高的PSM也是一种可能考虑的方法(图1 e)。我们对比了基于图1d的总体FDR和图1e的选择性PSM FDR,结果显示后者的FDR更严格。
图1丨de novo FDR计算
02验证从头测序的FDR
为了验证上述方法估计的FDR,首先将搜库的结果定义为true FDR。如果从头测序FDR与true FDR相匹配,则可以证明其是正确的,否则即为错误鉴定。但是,由于数据库搜索只需要匹配的到部分碎片离子即可识别多肽,因此一些多肽的二级谱中并没有完整的b/y离子。对于这些谱图,要求从头测序工具预测出准确的氨基酸序列是不公平的,因为生成decoy spectrums和随后的 FDR 计算主要基于碎片离子信息。因此,我们将正确的从头测序结果定义如下:如果从头测序多肽覆盖了谱图中真实碎片离子的Y%以上,则认为该多肽是“正确的”,Y%默认为90%。此参数允许用户根据对从头测序结果的期望进行调整,取决于数据类型、仪器、碎裂方式等。
理想情况下,Estimated FDR 等于true FDR(黑色虚线)。我们比较了几种不同的从头测序算法对同一批ABRF数据的FDR估算结果。图2是通过随机去除峰的方法比较的结果,可以看到不同的从头测序算法对生成的decoy spectrum的表现不尽相同,这些差异是不同算法和评分机制造成的。但可以为每种从头测序算法选择最合适的decoy spectrum百分比以获得更加接近true FDR的结果。图3是根据峰强度和肽质量数生成decoy spectrum的对比结果,与前面的发现一致,即该方法会产生更严格的FDR计算。
总体而言,不同的从头测序算法都各有不同,需要根据每个算法的模型和评分机制调整decoy spectrum的生成策略和参数。
理想情况下,Estimated FDR 等于true FDR(黑色虚线)。我们比较了几种不同的从头测序算法对同一批ABRF数据的FDR估算结果。图2是通过随机去除峰的方法比较的结果,可以看到不同的从头测序算法对生成的decoy spectrum的表现不尽相同,这些差异是不同算法和评分机制造成的。但可以为每种从头测序算法选择最合适的decoy spectrum百分比以获得更加接近true FDR的结果。图3是根据峰强度和肽质量数生成decoy spectrum的对比结果,与前面的发现一致,即该方法会产生更严格的FDR计算。
总体而言,不同的从头测序算法都各有不同,需要根据每个算法的模型和评分机制调整decoy spectrum的生成策略和参数。
 图2丨随机法生成decoy spectrums
图2丨随机法生成decoy spectrums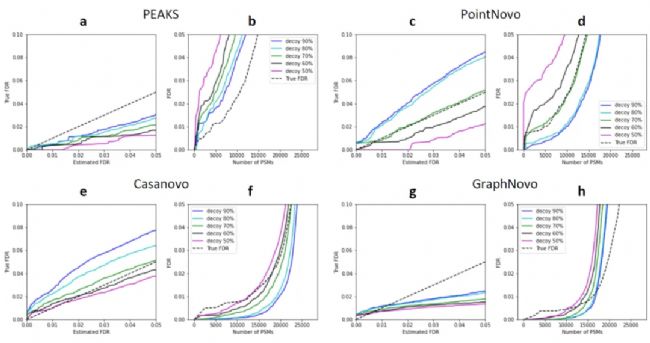 图3丨根据峰强度和肽质量数生成decoy spectrums
图3丨根据峰强度和肽质量数生成decoy spectrums03不同从头测序算法对胰酶酶切数据集的结果评估
首先,我们评估了五种多肽从头测序算法对ABRF数据集的分析结果,图4a表明这五种算法在该数据集上都表现整体良好,多肽、氨基酸和碎片离子水平的准确度分别达到 37-76%、68-88% 和 85-96%。PointNovo、Casanovo和GraphNovo的表现优于PEAKS®️ 常规de novo和Novor,在预期范围内,尤其是在多肽准确度方面。在氨基酸和碎片离子水平上,GraphNovo均优于其他算法,这要归功于其在对碎片离子特征模型建立方面的优势[2]。Casanovo的多肽序列准确度为76% ,但其氨基酸和碎片离子准确度比GraphNovo低约4-8%。这种差异表明,Casanovo可能在某些多肽上表现出色并能正确预测整个序列,但对于其他多肽,可能无法做出正确的预测。在相同FDR下,GraphNovo和Casanovo得到的PSM数量详尽,且均多于其他算法(图4b)。

图4丨ABRF标准胰酶酶切数据对比
然后,我们评估了以上几种算法对A.thaliana (PXD013658[3]) 的胰酶酶切数据集的表现。A. thaliana是一种与人类关系不太密切的物种,目前为止,尚未用于训练任何从头测序模型。正如预期,与ABRF数据集相比,从头测序结果的准确度大幅下降,多肽、氨基酸和碎片离子水平的准确度分别为27-47%、51-80%和68-88% (图4c、d)。其中GraphNovo表现最佳,Casanovo 受到了相当大的影响,多肽准确度下降了33%(76% 到43%),而其氨基酸和碎片离子准确度甚至低于所有其他算法。此A. thaliana数据集的结果表明深度学习模型在不同程度上偏向训练数据。值得关注的是,PEAKS®️常规de novo性能非常稳定,多肽和氨基酸准确度仅下降约4-5%。04非酶切和HLA数据集的从头测序评估
由于胰酶酶切数据集较多,现有算法模型可能存在偏向性,因此评估它们在非胰酶酶切数据上的表现至关重要。所以,我们对 Wang 等[4]发表的数据集进行了评估,其中包括Arg-C、Asp-N、Chymotrypsin、Glu-C、Lys-C和Lys-N的6种酶切数据。结果如图5 a-b所示,所有算法的整体性能都明显低于之前在胰蛋白酶数据上的测试结果。其中,GraphNovo 的多肽、氨基酸和碎片离子水平的准确度均远远优于其他算法。
我们进一步对 Wilhelm 等人的 HLA-I数据集(PXD021013[5])进行了评估。结果如图5c 所示,除Novor外,其他四种算法在该数据集上均表现良好,多肽准确率高达58-64%。这可能是因为 HLA-I类肽较短,长度为 8-12个氨基酸,因此更容易正确预测整个多肽序列。在 FDR 评估中,GraphNovo 和 Casanovo 报告的PSM数量最多,其次是 PointNovo 和 PEAKS®️ de novo。
我们进一步对 Wilhelm 等人的 HLA-I数据集(PXD021013[5])进行了评估。结果如图5c 所示,除Novor外,其他四种算法在该数据集上均表现良好,多肽准确率高达58-64%。这可能是因为 HLA-I类肽较短,长度为 8-12个氨基酸,因此更容易正确预测整个多肽序列。在 FDR 评估中,GraphNovo 和 Casanovo 报告的PSM数量最多,其次是 PointNovo 和 PEAKS®️ de novo。
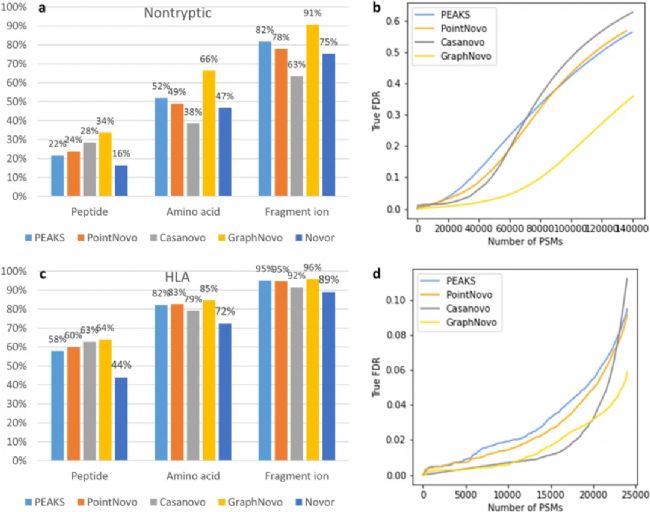
图5丨对非酶切( a、b)和 HLA 数据集(c、d )的从头测序结果的评估
为了进一步研究多肽长度对从头测序准确度的影响,我们重新评估了按多肽长度分布的 ABRF 数据集上的结果。如图6所示,准确率随着多肽长度的增加而降低。
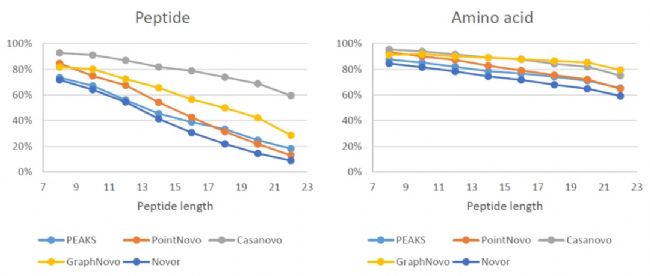
图6丨从头测序准确性随肽段长度的变化
05突变肽数据集的从头测序评估
我们在包含一万个突变肽的数据集上评估了从头测序工具。接下来,我们从 MassIVE-KB 数据库中随机挑选了一万条多肽,并且每个多肽中随机引入1-10个氨基酸突变,然后使用 Prosit[5]的2020 HCD模型预测数据集的谱图。图7a-b 显示,随着突变数量的增加,从头测序的准确性随之下降。在多肽水平上,Casanovo下降最明显 (45% to 24%),其次是 GraphNovo(33% to 23%)和 PEAKS®️ de novo (16% to 10%),而 PointNovo下降最少( 22% to 19%)。在氨基酸水平,GraphNovo和PointNovo约下降8%,而 Casanovo 和 PEAKS®️ de novo下降了约 20%。总体而言,Casanovo 似乎对突变数量最敏感,其次是 PEAKS®️、GraphNovo 和 PointNovo。与之前数据集结果类似,Casanovo肽段整体准确度较高,但在氨基酸准确度上均低于其他算法。
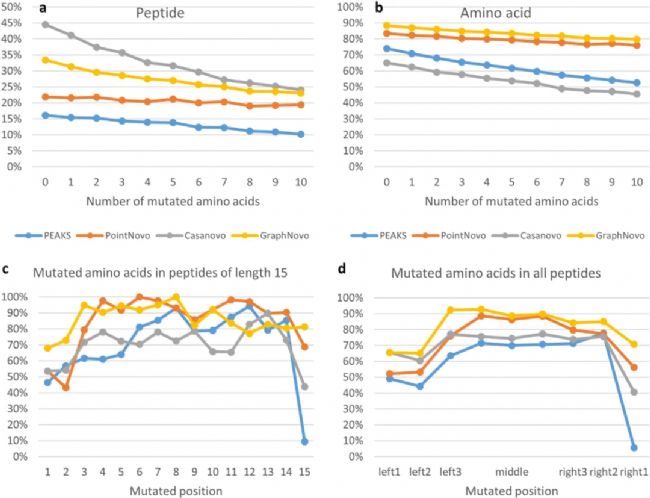
图7丨对突变肽模拟数据集的从头测序结果评估
我们进一步分析了突变氨基酸在肽段中的位置对从头测序结果的影响。如图7c所示,当突变发生在肽段的前后三个氨基酸位置时,对准确度影响较大,这是因为N端和C端的碎片离子缺失导致的,GraphNovo能够比其他算法更好地解决这个问题[2]。
06深度学习模型与多肽序列
基于深度学习的从头测序模型,最需要解决的问题是对训练数据中多肽序列的记忆。针对ABRF数据集的结果,我们统计了在MassIVE-KB训练集中存在的PSMs和不在MassIVE-KB中的PSMs的从头测序准确度。如图8a-b所示,不在MassIVE-KB中的PSMs的准确度大幅下降。但其通过搜库的方法得出的打分也偏低,这表明不在MassIVE-KB中的PSMs本身就是低质量谱图。因此,评估结果不是很准确。
为此,我们在相同实验条件下,重新采集了大肠杆菌、酵母和人类三个物种的质谱数据,搜库结果得到30-34K张PSMs,打分和多肽长度分布均比较一致。在MassIVE-KB中发现了大约96%的人类PSM,而大肠杆菌和酵母PSMs仅0-2%。然后,我们比较了这三个数据集的从头测序准确度,如图8c-d所示,PEAKS®️、PointNovo和 Novor对单个数据集的准确度几乎相同。而Casanovo和GraphNovo的结果显示,与人类样本相比,大肠杆菌和酵母的多肽准确率下降了3-4% ,表明深度学习算法存在一定的序列记忆和偏向性。
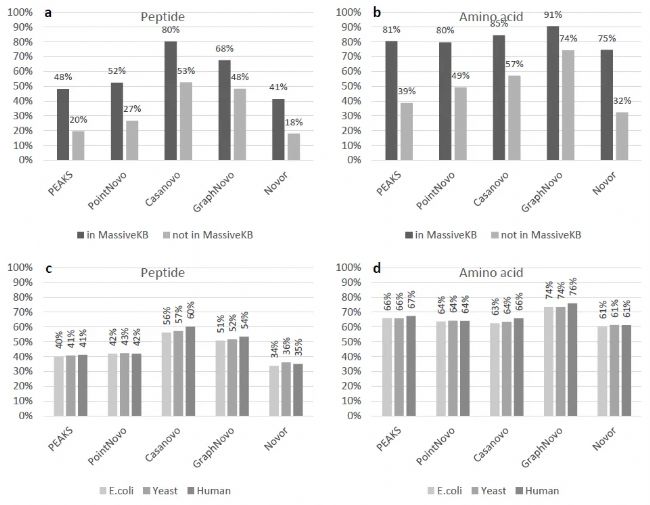 图8丨多肽训练记忆评估
图8丨多肽训练记忆评估原文链接
https://www.mcponline.org/article/S1535-9476(24)00139-7/fulltext
07小结
本研究提出了NovoBoard综合框架,用于综合评估多肽从头测序方法的性能、优缺点及其具体应用。重点关注基于深度学习的算法模型,以验证它们是否真正能够发现新序列,而不是过度拟合和记忆训练数据集,对未来的从头测序的广泛应用很有参考价值。但仍需不断优化,因为本研究只使用了Orbitrap的HCD DDA数据集,没有对低分辨和更多类型的数据进行训练和测试。此外,DIA 数据由于谱图的复杂性高,对于从头测序的挑战也更高,自DeepNovo- DIA [6]发表后,开启了对DIA数据的de novo分析,但也需要不断深入优化。
参考文献
1. Bittremieux, W. et al. Deep learning methods for de novo peptide sequencing. ChemRxiv (2024) doi:10.26434/chemrxiv-2024-l6wnt.
2. Mao, Z. Zhang, R. Xin, L. Mitigating the missing-fragmentation problem in de novo peptide sequencing with a two-stage graph-based deep learning model. Nature Machine Intelligence. 2023; 5:1250-1260.
3. Muntel, J. et al.Surpassing 10000 identified and quantified proteins in a single run by optimizing current LC-MS instrumentation and data analysis strategy. Mol Omics. 2019; 15:348-360.
4. Wang, D. et al. A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol. Syst. Biol. 15, e8503 (2019).
5. Wilhelm, M. et al. Deep learning boosts sensitivity of mass spectrometry-based immunopeptidomics. Nat. Commun. 12, 3346 (2021).
6. Tran NH, Li M et al. Deep learning enables de novo peptide sequencing from data-independent-acquisition mass spectrometry. Nat Methods. 2019 Jan;16(1):63-66.
2. Mao, Z. Zhang, R. Xin, L. Mitigating the missing-fragmentation problem in de novo peptide sequencing with a two-stage graph-based deep learning model. Nature Machine Intelligence. 2023; 5:1250-1260.
3. Muntel, J. et al.Surpassing 10000 identified and quantified proteins in a single run by optimizing current LC-MS instrumentation and data analysis strategy. Mol Omics. 2019; 15:348-360.
4. Wang, D. et al. A deep proteome and transcriptome abundance atlas of 29 healthy human tissues. Mol. Syst. Biol. 15, e8503 (2019).
5. Wilhelm, M. et al. Deep learning boosts sensitivity of mass spectrometry-based immunopeptidomics. Nat. Commun. 12, 3346 (2021).
6. Tran NH, Li M et al. Deep learning enables de novo peptide sequencing from data-independent-acquisition mass spectrometry. Nat Methods. 2019 Jan;16(1):63-66.

-扫码关注-
www.bioinfor.com (EN)
www.deepproteomics.cn(CN)
www.bioinfor.com (EN)
www.deepproteomics.cn(CN)
作为生物信息学的领军企业,BSI专注于蛋白质组学和生物药领域,通过机器学习和先进算法提供世界领先的质谱数据分析软件和蛋白质组学服务解决方案,以推进生物学研究和药物发现。我们通过基于AI的计算方案,为您提供对蛋白质组学、基因组学和医学的卓越洞见。旗下著名的PEAKS®️系列软件在全世界拥有数千家学术和工业用户,包括:PEAKS®️ Studio,PEAKS®️ Online,PEAKS®️ GlycanFinder, PEAKS®️ AB,DeepImmu®️ 免疫肽组发现服务和抗体综合表征服务等。
联系方式:021-60919891;sales-china@bioinfor.com
联系方式:021-60919891;sales-china@bioinfor.com



