
PEAKS QC功能和蛋白变化趋势设计结合实现最准确的TMT定量详解
PEAKS系列讲座火热报名中!!!
最后一讲将于3月13日(周三)上午10:00准时开启,由BSI产品经理李文婷分享“质谱数据定性、定量分析的全自动化与质量控制”的主题报告,点击“活动丨PEAKS系列讲座--蛋白组研究热点应用”,即可快速报名。
本论文的数据分析工作由Bioinformatics Solutions Inc.PEAKS Studio团队协助完成,成果已发表于J. Am. Soc. Mass Spectrom[1]。

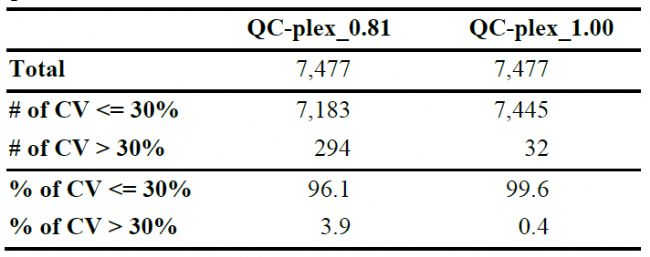
取4份野生型小鼠脑组织蛋白酶切肽段,按照0.81:1:1:0.81比例分别标记TMT10plex的127N、128N、129N和130N,所产生的质谱数据使用PEAKS Studio完成分析,PSM和Protein group的FDR均设置为1%,共得到75997 PSMs,46313 peptides,6776 protein groups,7477 proteins。同时,用PEAKS独有的Quality Control功能可以计算每一个PSM的CV值,结果如图1和表1,表明在蛋白质组学数据中,样本量越低,越容易产生更高的CVs。
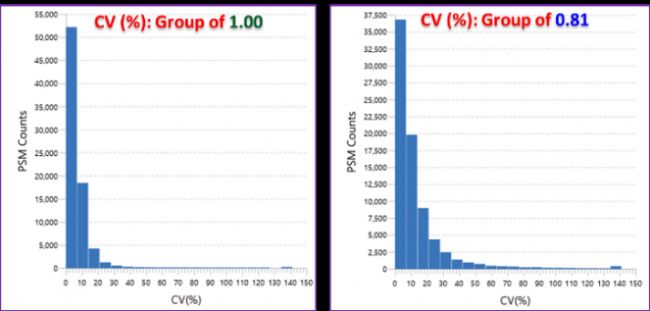
图1 两组QC-plex的CV值分布
表1 不同QC设置的定量蛋白统计
在PEAKS软件的TMT方法设置界面,可以添加Quality Control Replicate Channels,默认CV范围为≤30%(图2)。图3A-D中,紫色区域表示定量的假阳性结果,对比四种QC的设置方式可以看出,QC-plex越多,假阳性的定量就越少。
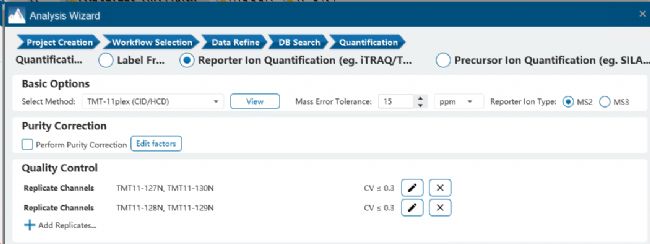
图2 PEAKS设置TMT Quality Control
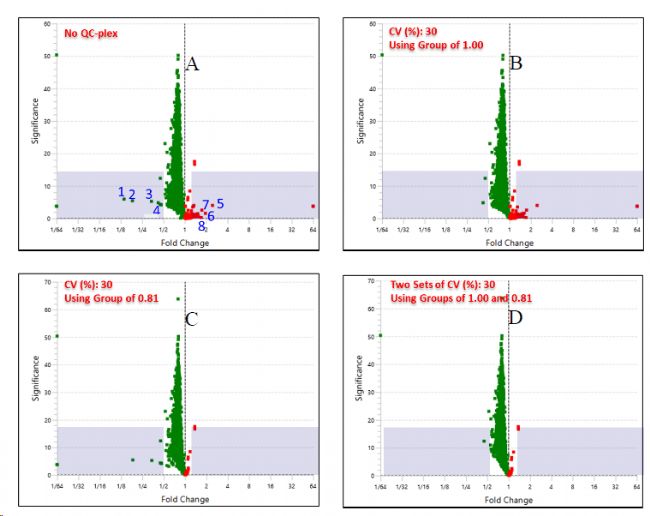
图3 QC-plex对定量假阳性的过滤
定量准确性与CV、unique peptides数量的关系
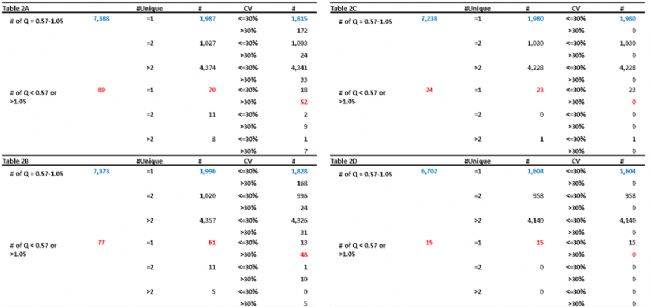
在表2中,进一步从unique peptide数量和CV方面比较了蛋白的定量准确性。当不使用QC-plex时,定量假阳性(Q<0.57 or >1.05)的蛋白有89个,分别设置QC-0.81、QC-1和QC-0.81&1后,定量假阳性的蛋白分别减少为77、24和15,并且也能看出unique peptide数量越少,越容易产生更高的CV,在应用了QC-plex后,即使unique peptide为1的蛋白,CV值也都在30%以下。
表2 不同QC设置的蛋白数统计
蛋白趋势变化实验设计
虽然上述实验验证了PEAKS QC功能对丰度较低且unique peptide数量少的蛋白定量的校正效果,但实际上,我们很难改变复杂生物学样本中提取出来的蛋白丰度,因此本研究又设计了图4所示的趋势变化实验(Trend design)的方案,从另一个角度实现TMT定量准确性的判断。该方案共包含5组小鼠:野生型小鼠组(WT)、无处理基因敲除小鼠组(non-Treated)、低剂量低亲和力分子处理基因敲除小鼠组(LowAffinity_LowDose)、低剂量高亲和力分子处理基因敲除小鼠组(HighAffinity_LowDose)、高剂量高亲和力分子处理基因敲除小鼠组(HighAffinity_HighDose),每组肽段使用不同channel的TMT试剂标记,然后等量混合进行质谱检测。

图4 不同分子亲和力富集及剂量差异设计
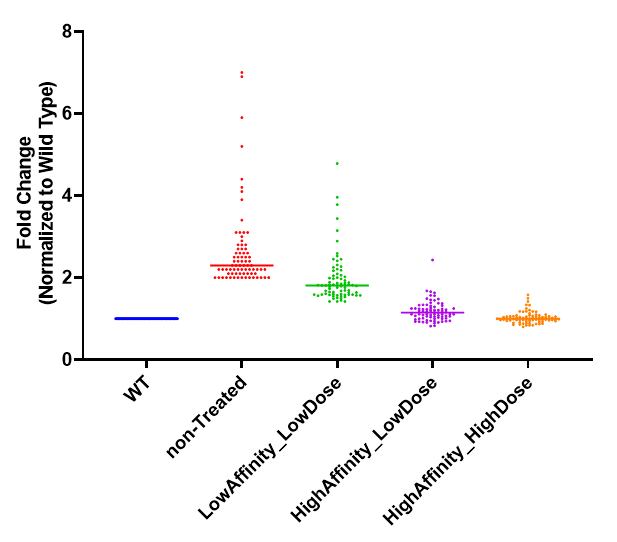
图5 不同处理组之间蛋白表达量的倍数变化
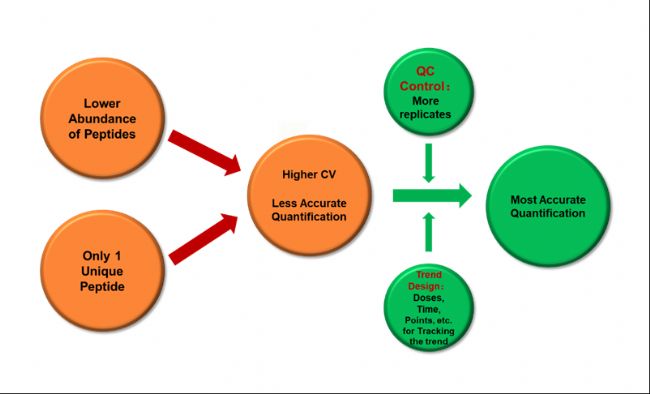
影响蛋白定量准确性的两个主要因素,一个是丰度低,一个是unique peptide数量过少,但我们并不能改变未知样品中存在的这两个客观问题,因此建议在实验过程中尽可能多做一些重复,通过QC-plex来计算CV,从而检验重现性。此外,结合Trend-design,可以达到最准确的TMT定量效果(图6)。
1. Xianyin Lai, Guihong Qi, Chris Kovach, Yaming Wang, Isaiah Clark, Keyue Chen, Zhixiang Yang, Nick Babb, Forest Andrews, Ross Fellows, Baozhen Shan, Weiwu Chen, Tom Yang, and Wenting Li. Journal of the American Society for Mass Spectrometry . DOI: 10.1021/jasms.3c00346.

-扫码关注-
www.bioinfor.com (EN)
www.deepproteomics.cn(CN)
作为生物信息学的领军企业,BSI专注于蛋白质组学和生物药领域,通过机器学习和先进算法提供世界领先的质谱数据分析软件和蛋白质组学服务解决方案,以推进生物学研究和药物发现。我们通过基于AI的计算方案,为您提供对蛋白质组学、基因组学和医学的卓越洞见。旗下著名的PEAKS系列软件在全世界拥有数千家学术和工业用户,包括:PEAKS Studio,PEAKS Online,PEAKS GlycanFinder, PEAKS AB及抗体综合表征服务等。
联系方式:021-60919891;sales-china@bioinfor.com



