
NetMHCIIpan4.3准确预测HLA-Il类抗原在所有基因座呈递详解
背景介绍

使用到的免疫肽组学数据集概览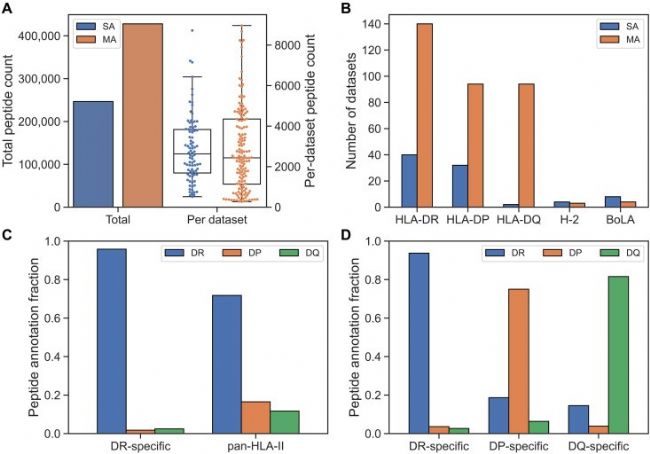
图1 数据集概览
DP数据和反向结合模式对于预测性能的影响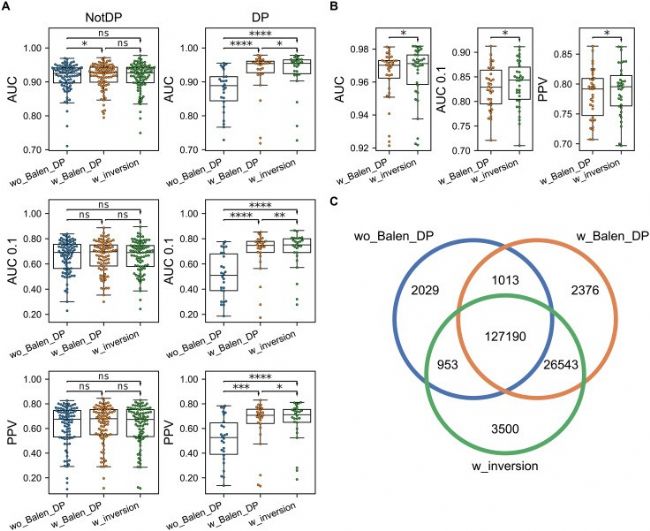
图2 HLA-DP预测性能评估
反转结合基序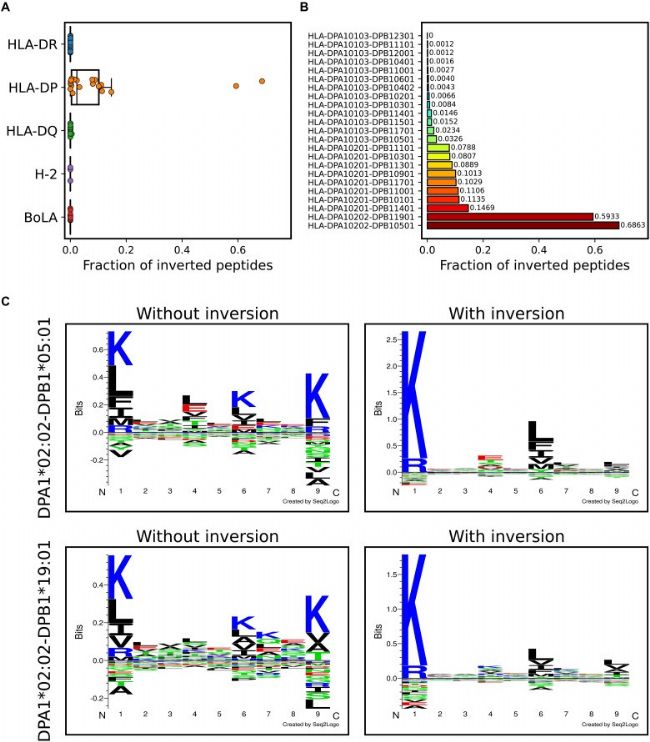
图3肽反转模型使得DP基序解卷积更准确
解卷积结果与预测基序的关联度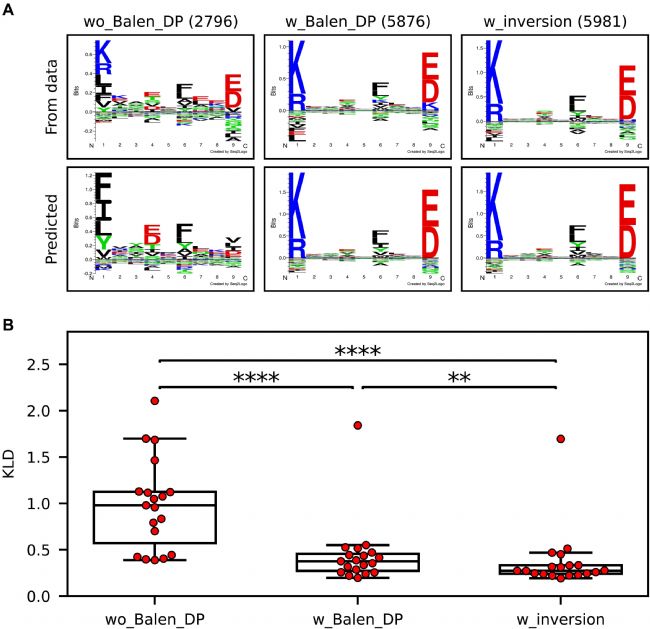
HLA-DP分子覆盖率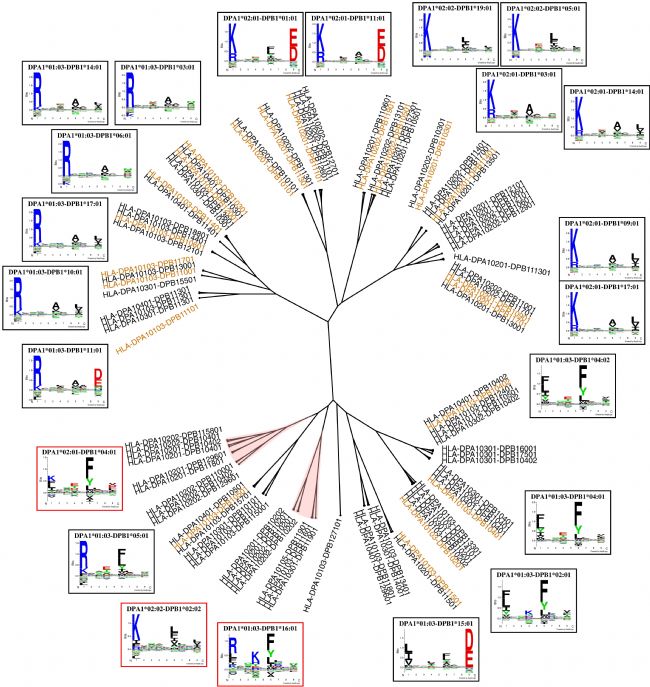
预测方法模型的其他性能测试
01
使用包括多肽编码在内的反转模型对该方法进行了重新训练,观察到在所有三个HLA-II基因座 (DR、DP和DQ)中性能显著提高, HLA-DQ改善最多(AUC、AUC 0.1和PPV分别增加3.5、9.4和5.8个百分点,见图S9)。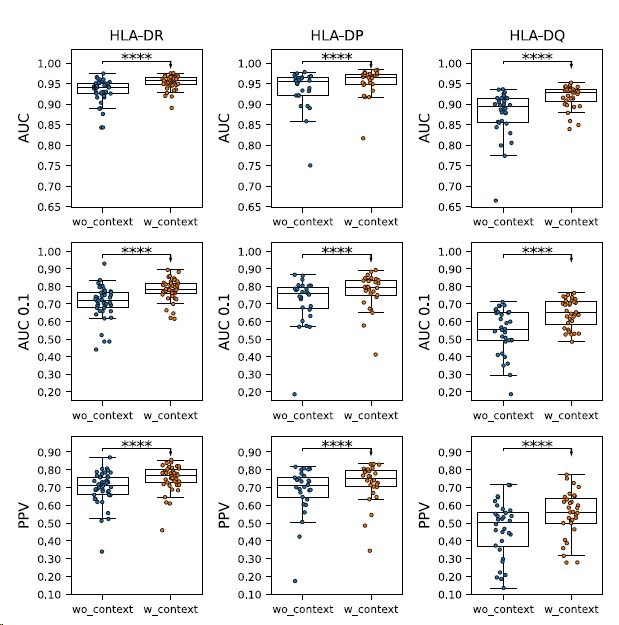
图S9 多肽编码模型重训练
图S10
03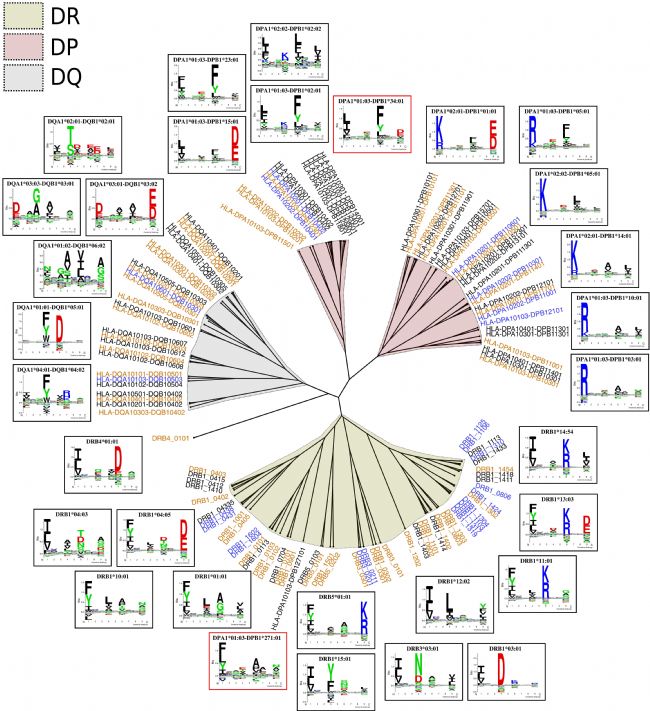
图7 HLA- DR、HLA- DP和HLA- DQ分子的特异性树
04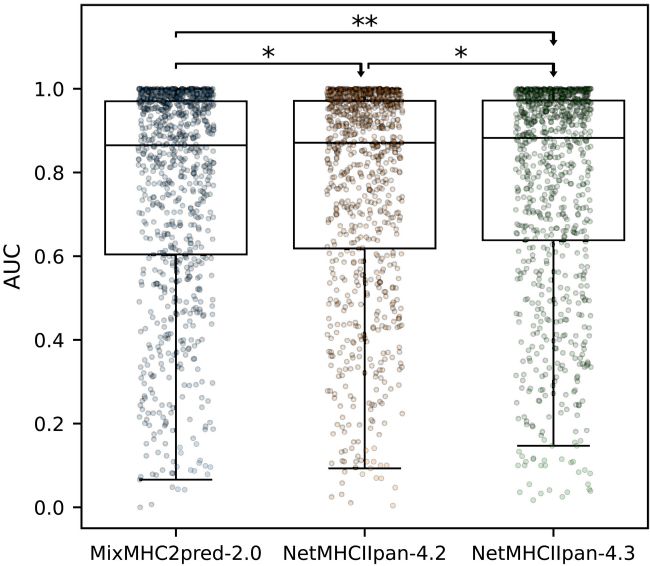
图8 CD4+表位基准测试
总结
若您想深入了解PEAKS软件在免疫肽组分析中的应用,欢迎点击“阅读原文”提交您的咨询信息!
参考文献
主要组织相容性复合体(MHC)II类分子,也被称为人类白细胞抗原(HLA)II类分子,在特异性抗原呈递细胞表面表达,通过向CD4+T细胞呈递抗原肽,在免疫系统功能中起关键作用[1,2]。从结构上看,这些分子是由三个不同的基因座(HLA-DR, HLA-DP, HLA-DQ)编码的,并由α链和β链组成的异源二聚体,是人类基因组中多态性最高的基因之一[3]。这些多态性大多聚集在由α和β链形成的肽结合区域周围,从而产生广泛的肽结合特异性。HLA研究中,模拟免疫反应激活的过程,就要经过蛋白酶体对抗原(蛋白)的剪切预测、肽段转运、肽段和MHC-I结合亲和力预测以及T细胞识别预测等几个重要步骤。准确预测人类白细胞抗原(HLA)II类分子的抗原呈递对于合理开发针对CD4+T细胞活化的免疫疗法和疫苗至关重要。
2023年11月24日,Science Advances(IF=13.6)发表了NetMHCIIpan的最新算法文章“Accurate prediction of HLA class II antigen presentation across all loci using tailored data acquisition and refined machine learning”。作者将覆盖所有三个基因座的大规模高质量免疫肽组学数据集整合到NetMHCIIpan机器学习框架中,并应用最新版本的NNAlign_MA方法,将正向与反向的肽结合模式(正向结合从N端到C端,反向为C端到N端方向结合)预测纳入方法训练模型,从而缩小DR和DQ/DP之间的差距。利用这种方法,研究了HLA-DR、HLA-DQ和HLA-DP的预测性能,以及反向结合模式的预测如何影响基序解卷积,扩大了HLA覆盖范围。最终,NetMHCIIpan-4.3在所有HLA II类同种异型抗原中实现了高精度和分子覆盖率。其中,免疫肽组学质谱数据的分析使用PEAKS Studio 11完成。
使用到的免疫肽组学数据集概览
作者整合了在NetMHCIIpan-4.2模型训练中使用到的免疫肽组学数据集,从图1A可以看出大多数训练集的数据来自于MA(multi-allelic)组学数据。图1B显示了每个基因座的SA(single-allelic)和MA数据集数量,表明Balen_DP[4-6]数据的引入使得HLA-DP具有与HLA-DR相似数量的SA数据集,HLA-DQ的SA数据较少。图1C和D展示了两个样本在HLA基序解卷积中分配给DR、DP和DQ的肽的比例,其中一个样本使用传统的两步免疫沉淀方案处理(即先后使用泛DR抗体和泛II类抗体处理),第二个样本使用单独的DR、DQ和DP位点特异性抗体处理。图1C显示泛II类抗体的DP和DQ特异性较差,导致这两个位点的肽产量非常低。相反,图1D展示了应用三种位点特异性抗体产生的高特异肽。
图1 数据集概览
DP数据和反向结合模式对于预测性能的影响
基于上述数据集,大幅提高了DP和DQ肽的产量,然后使用NNAlign_MA框架训练HLA抗原呈递的预测模型[7]。在模型训练之前,对质谱数据进行预处理,并用随机生成的天然阴性对照肽进行富集。同时预测结合核心偏移和肽配体的正向与反向结合。然后,训练了三个初始预测模型来评估Balen_DP数据的影响:一个不包含Balen数据和没有反向肽结合(wo_Balen_DP),一个包含Balen数据和没有反向肽结合(w_Balen_DP),一个包含Balen数据和使用反向肽结合 (w_inversion)。最后,使用每个分子和每个样品的交叉验证对这些方法进行评估。
通过AUC(area under ROC curve)、AUC0.1(FDR为10%时的area under ROC curve)和PPV(positive predictive value)三个指标来评估预测性能。图2A表明Balen_DP数据中包含的信息对该方法学习其他基因座的特异性的影响有限,而对DP的预测都有显著的性能提高。图2B进一步表明,包含肽反向结合的模型也会显著改善DP的预测性能。并且,在所有注释的163604个DP肽中,有78%(127190条)的DP肽是三个模型共同注释到(图2C),表明这些方法模型对DP配体的高度识别。
通过AUC(area under ROC curve)、AUC0.1(FDR为10%时的area under ROC curve)和PPV(positive predictive value)三个指标来评估预测性能。图2A表明Balen_DP数据中包含的信息对该方法学习其他基因座的特异性的影响有限,而对DP的预测都有显著的性能提高。图2B进一步表明,包含肽反向结合的模型也会显著改善DP的预测性能。并且,在所有注释的163604个DP肽中,有78%(127190条)的DP肽是三个模型共同注释到(图2C),表明这些方法模型对DP配体的高度识别。
图2 HLA-DP预测性能评估
反转结合基序
图3A显示了反向肽在HLA分子中不同位点的百分比分布。由此,发现肽反转几乎只发生在HLA-DP上。当观察每个DP分子的反转百分比时(图3B),看到含有5%以上肽反转的分子都具有DPA1*02:01或DPA1*02:02的α链。此外,小于5%肽反转的分子都具有相同的DPA1*01:03 α链。这表明,HLA-DP α链是反向肽结合模式的主要决定因素。这些观察结果与近期的一些研究趋势一致,但作者的方法比先前研究更多地预测到了DPB1*03:01反向肽。另外,图3C展示了经过和没有经过反转训练模型的DPA1*02:02-DPB1*05:01和DPA1*02:02-DPB1*19:01的基序,对于没有反转的方法,识别出的基序是围绕中心位置镜像分布的,K和R同时存在于P1和P9。相反,对于反转训练的模型,由于考虑了双重结合模式,使得基序更加清晰,K和R偏好仅在P1处存在。进一步证明了纳入反向肽的模型后对DP基序解卷积的改善。
图3肽反转模型使得DP基序解卷积更准确
解卷积结果与预测基序的关联度
接下来,作者研究了Balen_DP数据中的19个HLA-DP分子,通过比较解卷积获得的结合基序与基于随机天然肽的预测基序之间的相关性,评估该训练模型对MS数据中结合基序的学习能力。图4A展示了DPA1*02:01-DPB1*01:01的数据,在不同的训练模型中得出的解卷积基序总体上一致。但是从预测的结果上看,不使用Balen_DP数据的模型无法完全学习正确的基序。此外,反转方法无论是在预测,还是解卷积到的基序,一致性都很高,可以将“K”定位在P1而不是P9。
然后,基于Balen_DP数据中所有样本的交叉验证预测和上述得分最高的随机天然肽,为Balen_DP数据中的每个分子构建了位置特异性频率矩阵(PSFMs)。这个KLD度量可以解释为两个分子结合基序之间的“距离”,其中较低的值表明更多相似的基序。分析结果如图4B所示,包含反转方法的模型KLD值显著低于未包含反转的方法。另外观察到,在大多数情况下,具有相同β链的分子对具有相似的基序,这表明β链是DP分子的主要特异性定义元件。
然后,基于Balen_DP数据中所有样本的交叉验证预测和上述得分最高的随机天然肽,为Balen_DP数据中的每个分子构建了位置特异性频率矩阵(PSFMs)。这个KLD度量可以解释为两个分子结合基序之间的“距离”,其中较低的值表明更多相似的基序。分析结果如图4B所示,包含反转方法的模型KLD值显著低于未包含反转的方法。另外观察到,在大多数情况下,具有相同β链的分子对具有相似的基序,这表明β链是DP分子的主要特异性定义元件。
图4 观察到的和预测到的基序之间的对应关系
HLA-DP分子覆盖率
经过前面的工作,基本验证了该方法对HLA II类抗原较好的预测能力,接下来作者又对HLA-DP分子的覆盖范围做了探究。通过考虑在不超过0.05的距离内发现的DP分子参考集与肽覆盖分子的比例来估计功能覆盖范围,从分析中发现包含Balen_DP数据的模型功能性DP覆盖率显著增加。接下来,使用反转的方法,基于MHCCluster方法构建了一个DP特异性树[8]。简而言之,基于伪序列,167个流行的DP分子被减少到95个具有独特特异性的分子[9]。然后,根据大量随机天然肽的预测分数之间的相关性估计分子之间的距离,得到如图5所示的树。从特异性树中观察到该模型具有不同DP特异性的广泛覆盖范围,大多数分支至少具有一个肽覆盖分子(具有至少50个可信肽注释的分子)。然而,一些分支机构被覆盖率较低。其中一个分支包括DPA1*02:01DPB1*04:01,该分支的基序没有通过前面描述的预测方法正确学习。该分子存在于训练数据中的7个样本中,均为异杂合子。在所有这些样本中,该分子被分配的DP注释少于5%,导致有效肽计数为0。这种肽注释的缺乏可能是生物学上的原因,也可能是由于缺乏高质量的分子数据而导致该方法没有充分学习到分子的特异性。
在上述分析的基础上,利用DP特异性免疫沉淀法从表达DPA1*01:03 - DPB1 *16:01、DPA1*02:01-DPB1*04:01和DPA1*02:02 - dDPB1 *02:02的DP纯合子细胞系中获得免疫肽组学质谱数据。FDR小于1的肽段数分别为2423、1797和2428。在去除翻译后修饰和冗余肽后,每个样本中独特的12- 21-mer肽的数量分别减少到1550,1259和1502,然后使用反转重新训练的方法,以评估它们对DP基序解卷积和分子覆盖的影响。
结果显示,重新训练的方法能够从每个单独的数据集中注释678(53.8%)、715(47.6%)和1062(68.5%)个百分位等级小于20的多肽,分别指向DPA1*02:01-DPB1*04:01、DPA1*02:02-DPB1*02:02和DPA1*01:03-DPB1*16:01,其基序如图6A所示。剩余的多肽是主要分配给HLA-DR和HLA-DQ共流出肽段。据预测,DPA1*02:02- DPB1 *02:02的反转结合肽比例很高(25.3%),而反转肽在P4位点对组氨酸有偏好(图6B)。此外,比较了DP注释肽的长度分布,证实了15-mer肽的正态分布,这与大多数HLA I类(包括其他DP分子)的长度偏好一致(图6C)。将包含和不包含这些数据集的模型进行比较,正如预期,具有肽覆盖的DP分子数增加了(24到27),使得167个DP分子的覆盖范围扩大到131个(早期模型的167个分子覆盖116个),人群覆盖率扩大到96%,如图6D所示。
在上述分析的基础上,利用DP特异性免疫沉淀法从表达DPA1*01:03 - DPB1 *16:01、DPA1*02:01-DPB1*04:01和DPA1*02:02 - dDPB1 *02:02的DP纯合子细胞系中获得免疫肽组学质谱数据。FDR小于1的肽段数分别为2423、1797和2428。在去除翻译后修饰和冗余肽后,每个样本中独特的12- 21-mer肽的数量分别减少到1550,1259和1502,然后使用反转重新训练的方法,以评估它们对DP基序解卷积和分子覆盖的影响。
结果显示,重新训练的方法能够从每个单独的数据集中注释678(53.8%)、715(47.6%)和1062(68.5%)个百分位等级小于20的多肽,分别指向DPA1*02:01-DPB1*04:01、DPA1*02:02-DPB1*02:02和DPA1*01:03-DPB1*16:01,其基序如图6A所示。剩余的多肽是主要分配给HLA-DR和HLA-DQ共流出肽段。据预测,DPA1*02:02- DPB1 *02:02的反转结合肽比例很高(25.3%),而反转肽在P4位点对组氨酸有偏好(图6B)。此外,比较了DP注释肽的长度分布,证实了15-mer肽的正态分布,这与大多数HLA I类(包括其他DP分子)的长度偏好一致(图6C)。将包含和不包含这些数据集的模型进行比较,正如预期,具有肽覆盖的DP分子数增加了(24到27),使得167个DP分子的覆盖范围扩大到131个(早期模型的167个分子覆盖116个),人群覆盖率扩大到96%,如图6D所示。
图5 HLA-DP特异性分布树
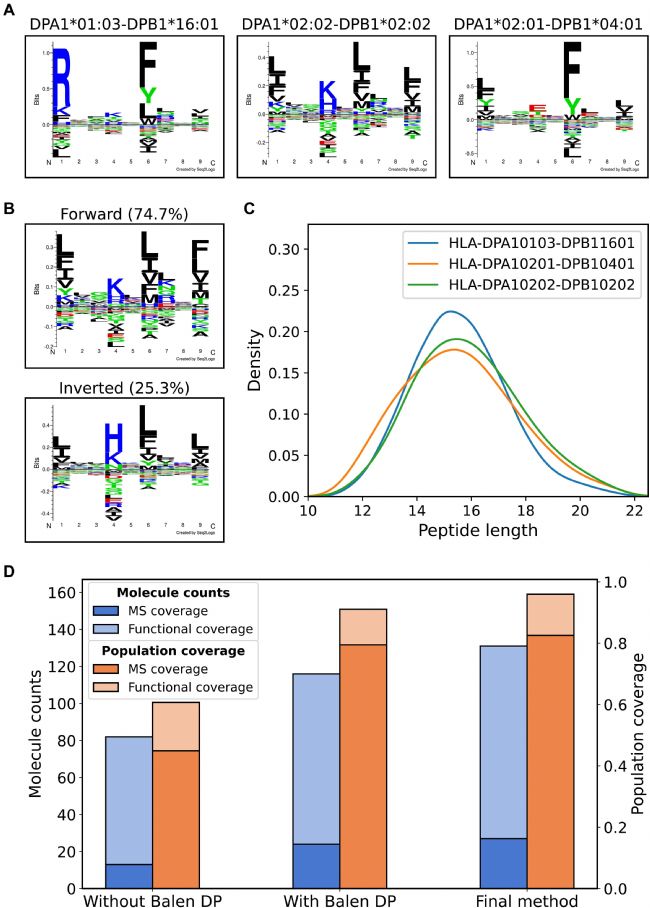
图6 整合额外DP数据集后的分子覆盖率
图6 整合额外DP数据集后的分子覆盖率
预测方法模型的其他性能测试
使用包括多肽编码在内的反转模型对该方法进行了重新训练,观察到在所有三个HLA-II基因座 (DR、DP和DQ)中性能显著提高, HLA-DQ改善最多(AUC、AUC 0.1和PPV分别增加3.5、9.4和5.8个百分点,见图S9)。
图S9 多肽编码模型重训练
02
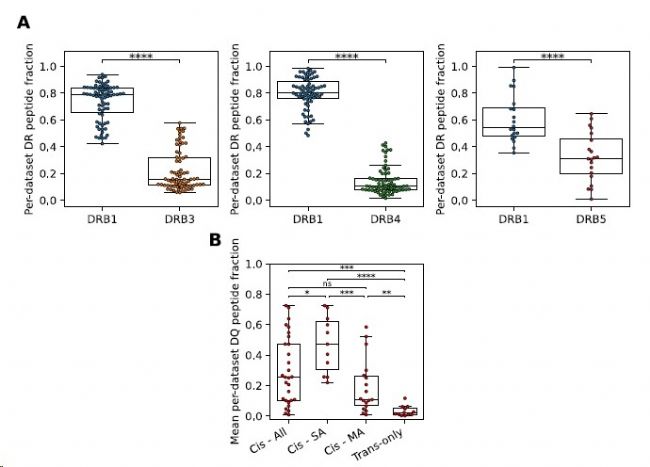
使用上述方法模型进一步分析对HLA-DR和HLA-DQ的预测能力。通过绘制每对分子(即DRB1与DRB3, DRB1与DRB4, DRB1与DRB5)的DR肽注释分数在每个数据集中的分布,发现在含有DRB1和DRB5的样品中,DRB5总体上具有较高的肽贡献(见图S10A)。另一方面,DRB4的贡献最低,而DRB3的贡献不太一致,说明DRB3基因具有更多的多态性。这些结果与Kaabinejadian等人[10]的发现非常吻合,再次说明了在基序解卷积过程中包含完整HLA-DR分型以准确表征DR配体的重要性。此外,分析了DQ杂合数据集的基序解卷积以及HLA-DQ和β链配对在形成免疫肽丘中的作用。如图S10B所示,反式组合在所有DQ-杂合数据集中的贡献始终较低,DQ-MA数据集中发现的顺式变异的贡献明显高于反式变异。然而,我们观察到DQ-SA数据集中存在的顺式变异比DQ-MA数据集中存在的顺式变异的总体贡献更高,表明对这些分子的潜在偏好。
图S10
03
构建了一个结合HLA- DR、HLA- DP和HLA- DQ分子的特异性树以评估整个HLA II类特异性覆盖。基于伪序列之间的相似性,得到53个具有独特特异性的DR分子,40个DP分子和24个DQ分子。然后,使用MHCCluster方法构建这些分子的总体特异性树。其结果如图7所示。总体来看,每个位点上的分子被分在一组,形成定义明确的簇。也有少数例外,DRB4*01:01单独定位在DQ分支附近,DPA1*01:03-DPB1*271:01与一组DR分子聚集在一起。后者很可能是由于这个DP分子在肽覆盖和伪序列距离方面都没有被目前的方法覆盖。
图7 HLA- DR、HLA- DP和HLA- DQ分子的特异性树
04
作为对NetMHCIIpan-4.3的最终验证,对其在CD4+表位鉴定中的性能进行了基准测试。结果如图8所示,NetMHCIIpan-4.3显著优于MixMHC2pred-2.0和NetMHCIIpan-4.2 。
图8 CD4+表位基准测试
经过对多重数据多种模型的优化与验证,最终将该预测模型整合形成NetMHCIIpan-4.3工具,有效地缩小了HLA- DP/HLA- DQ和HLA- DR之间的性能差距,从而增强了所有三个HLA II类位点的基序特征。借助NetMHCIIpan-4.3,HLA II类分子的特异性难题可以得到解决,有望拓宽我们对所有HLA II类在感染性和自身免疫性疾病中启动细胞免疫的分子作用机制,而不仅仅是HLA- DR。
若您想深入了解PEAKS软件在免疫肽组分析中的应用,欢迎点击“阅读原文”提交您的咨询信息!
参考文献
[1] S. Tsai, P. Santamaria, MHC class II polymorphisms, autoreactive T-cells, and autoimmunity. Front. Immunol. 4, 321 (2013).
[2] M. T. Arango, C. Perricone, S. Kivity, E. Cipriano, F. Ceccarelli, G. Valesini, Y. Shoenfeld, HLA-DRB1 the notorious gene in the mosaic of autoimmunity. Immunol. Res. 65, 82–98 (2017).
[3] M. Van Lith, R. M. McEwen-Smith, A. M. Benham, HLA-DP, HLA-DQ, and HLA-DR have different requirements for invariant chain and HLA-DM. J. Biol. Chem. 285, 40800–40808 (2010).
[4] P. van Balen, M. G. D. Kester, W. De Klerk, P. Crivello, E. Arrieta-Bolaños, A. H. De Ru,I. Jedema, Y. Mohammed, M. H. M. Heemskerk, K. Fleischhauer, P. A. Van Veelen,
J. H. F. Falkenburg, Immunopeptidome analysis of HLA-DPB1 allelic variants reveals new functional hierarchies. J. Immunol. 204, 3273–3282 (2020).
[5] S. Klobuch, J. J. Lim, P. van Balen, M. G. D. Kester, W. de Klerk, A. H. de Ru, C. R. Pothast, I. Jedema, J. W. Drijfhout, J. Rossjohn, H. H. Reid, P. A. van Veelen, J. H. F. Falkenburg, M. H. M. Heemskerk, Human T cells recognize HLA-DP–bound peptides in two orientations. Proc. Natl. Acad. Sci. U.S.A. 119, e2214331119 (2022).
[6] A. Laghmouchi, M. G. D. Kester, C. Hoogstraten, L. Hageman, W. de Klerk, W. Huisman, E. A. S. Koster, A. H. de Ru, P. van Balen, S. Klobuch, P. A. van Veelen, J. H. F. Falkenburg, I. Jedema, Promiscuity of peptides presented in HLA-DP molecules from different immunogenicity groups is associated with T-cell cross-reactivity. Front. Immunol. 13, 831822 (2022).
[7] B. Alvarez, B. Reynisson, C. Barra, S. Buus, N. Ternette, T. Connelley, M. Andreatta, M. Nielsen, NNAlign_MA; MHC peptidome deconvolution for accurate MHC binding motif characterization and improved T-cell epitope predictions. Mol. Cell. Proteomics 18, 2459–2477 (2019).
[8] M. C. F. Thomsen, C. Lundegaard, S. Buus, O. Lund, M. Nielsen, MHCcluster, a method for functional clustering of MHC molecules. Immunogenetics 65, 655–665 (2013).
[9] E. Karosiene, M. Rasmussen, T. Blicher, O. Lund, S. Buus, M. Nielsen, NetMHCIIpan-3.0, a common pan-specific MHC classII prediction method including all three human MHC class II isotypes, HLA-DR, HLA-DP and HLA-DQ. Immunogenetics 65, 711–724 (2013).
[10] S. Kaabinejadian, C. Barra, B. Alvarez, H. Yari, W. H. Hildebrand, M. Nielsen, Accurate MHC motif deconvolution of immunopeptidomics data reveals a significant contribution of
DRB3, 4 and 5 to the Total DR Immunopeptidome. Front. Immunol. 13, 835454 (2022).
[2] M. T. Arango, C. Perricone, S. Kivity, E. Cipriano, F. Ceccarelli, G. Valesini, Y. Shoenfeld, HLA-DRB1 the notorious gene in the mosaic of autoimmunity. Immunol. Res. 65, 82–98 (2017).
[3] M. Van Lith, R. M. McEwen-Smith, A. M. Benham, HLA-DP, HLA-DQ, and HLA-DR have different requirements for invariant chain and HLA-DM. J. Biol. Chem. 285, 40800–40808 (2010).
[4] P. van Balen, M. G. D. Kester, W. De Klerk, P. Crivello, E. Arrieta-Bolaños, A. H. De Ru,I. Jedema, Y. Mohammed, M. H. M. Heemskerk, K. Fleischhauer, P. A. Van Veelen,
J. H. F. Falkenburg, Immunopeptidome analysis of HLA-DPB1 allelic variants reveals new functional hierarchies. J. Immunol. 204, 3273–3282 (2020).
[5] S. Klobuch, J. J. Lim, P. van Balen, M. G. D. Kester, W. de Klerk, A. H. de Ru, C. R. Pothast, I. Jedema, J. W. Drijfhout, J. Rossjohn, H. H. Reid, P. A. van Veelen, J. H. F. Falkenburg, M. H. M. Heemskerk, Human T cells recognize HLA-DP–bound peptides in two orientations. Proc. Natl. Acad. Sci. U.S.A. 119, e2214331119 (2022).
[6] A. Laghmouchi, M. G. D. Kester, C. Hoogstraten, L. Hageman, W. de Klerk, W. Huisman, E. A. S. Koster, A. H. de Ru, P. van Balen, S. Klobuch, P. A. van Veelen, J. H. F. Falkenburg, I. Jedema, Promiscuity of peptides presented in HLA-DP molecules from different immunogenicity groups is associated with T-cell cross-reactivity. Front. Immunol. 13, 831822 (2022).
[7] B. Alvarez, B. Reynisson, C. Barra, S. Buus, N. Ternette, T. Connelley, M. Andreatta, M. Nielsen, NNAlign_MA; MHC peptidome deconvolution for accurate MHC binding motif characterization and improved T-cell epitope predictions. Mol. Cell. Proteomics 18, 2459–2477 (2019).
[8] M. C. F. Thomsen, C. Lundegaard, S. Buus, O. Lund, M. Nielsen, MHCcluster, a method for functional clustering of MHC molecules. Immunogenetics 65, 655–665 (2013).
[9] E. Karosiene, M. Rasmussen, T. Blicher, O. Lund, S. Buus, M. Nielsen, NetMHCIIpan-3.0, a common pan-specific MHC classII prediction method including all three human MHC class II isotypes, HLA-DR, HLA-DP and HLA-DQ. Immunogenetics 65, 711–724 (2013).
[10] S. Kaabinejadian, C. Barra, B. Alvarez, H. Yari, W. H. Hildebrand, M. Nielsen, Accurate MHC motif deconvolution of immunopeptidomics data reveals a significant contribution of
DRB3, 4 and 5 to the Total DR Immunopeptidome. Front. Immunol. 13, 835454 (2022).
标签:
免疫肽分析



