
PEAKS Online在大规模DDA数据集构建谱图库及DIA数据分析性能详解
使用已发表文献中的数据集[1]表明PEAKS Online在对大规模DDA数据集构建谱图库以及随后使用该库对DIA数据分析方面的性能。通过多CPU/GPU集群和服务器,PEAKS Online可以极大地缩短大型项目的分析时间。
背景介绍
数据依赖采集(DDA)和数据非依赖采集(DIA)方法是蛋白质组学串联质谱(MS2)的两种数据采集方式。在DDA模式下,质谱仪从MS1扫描中选择固定数量的响应较强的母离子用于MS2分析。相反,在DIA中,在MS2采集过程中,在选定的质荷比(m/z)窗口内的所有母离子都会被碎裂。虽然DDA一直是定性和定量蛋白质组学中最常用的方法,但DIA因为对低信号峰的检测和可重复性而受到欢迎。然而,由于DIA的一张MS2谱图中的碎片离子,可能是由多个母离子碎裂产生,因此母离子与其碎片离子之间的关联就变得很重要。
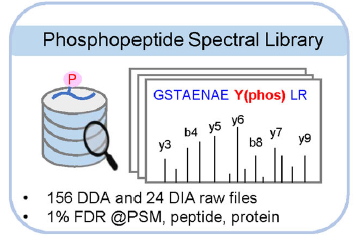
由于在DIA分析中产生的MS2谱图复杂度高,构建一个高质量和高覆盖率的谱图库是辅助DIA数据解析的首选。谱图库通常是在同一仪器上对所有样品的等量混合肽段进行多次分馏后通过DDA采集获得,然后根据物种蛋白质序列数据库进行检索完成定性,生成谱图库。对复杂DIA MS2谱图进行解卷积后,根据碎片离子和母离子的profile特征,将单个碎片离子分配到对应的母离子上。然后将MS2谱中的碎片离子峰与谱图库中的肽碎片离子相匹配,并结合m/z误差和保留时间(RT)等其他特征完成肽段鉴定[2]。
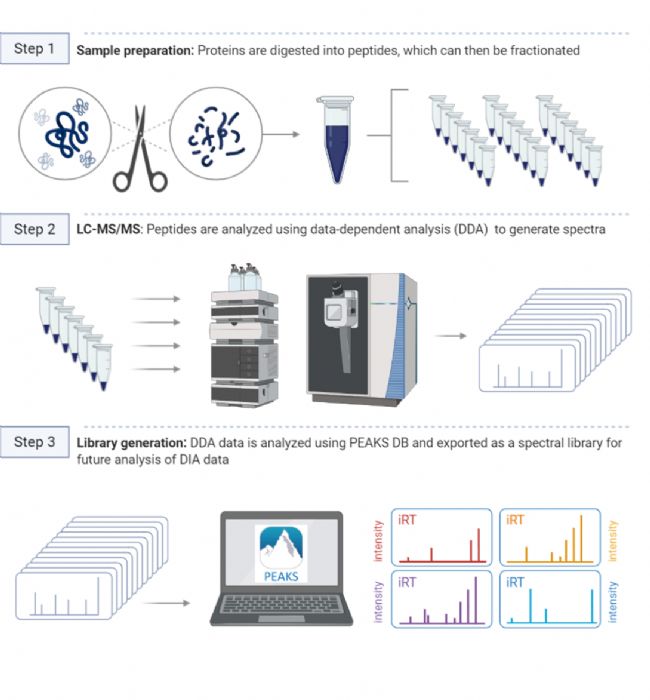
图1.如何构建谱图库
方法
实验设计
为了构建谱图库,对4株细胞系和6个肿瘤组织进行裂解、酶切和高pH反相色谱(HpRP)组份分离,然后通过StageTip用Fe-IMAC富集磷酸化肽段,在DDA模式下进行数据采集(图2a)。另外,将合成的与肺癌有关驱动基因表达的磷酸化肽段也纳入到谱图库中,以提高库的覆盖范围。挑选2个细胞系的样本进行DIA数据采集,用于谱图库分析。
(a) Sample Preparation
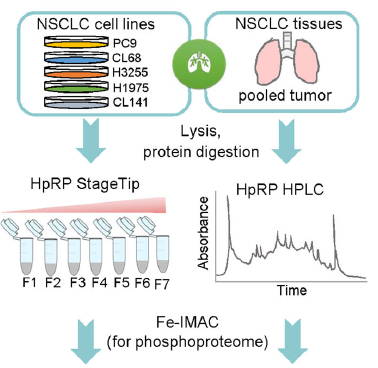
Fig 2. DDA和DIA实验
数据分析
(b) Spectral L ibrary
(c) DIA Analysis
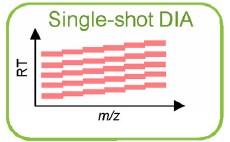
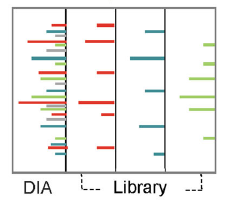
Fig 2. DDA和DIA实验
使用DDA数据集构建谱图库
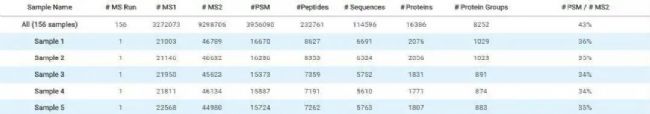
Fig 3. PEAKS Online中156个DDA数据的搜库结果
DIA数据的谱图库检索分析
谱图库检索结果包含DIA的MS2谱与谱图库的精确匹配肽段,如图4所示。
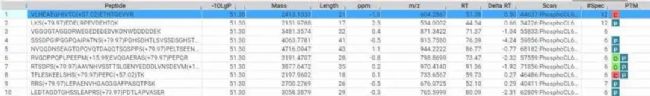
图4. 谱图库检索结果的肽段列表
对于每一条鉴定到的肽段,母离子和强度最高的几个碎片离子的XIC图都会可视化展示(图5)。此外,还提供了DIA MS2谱图中碎片离子峰与谱图库中对应肽段碎片离子峰的镜像对比图和离子列表(图6),以便进一步评估。
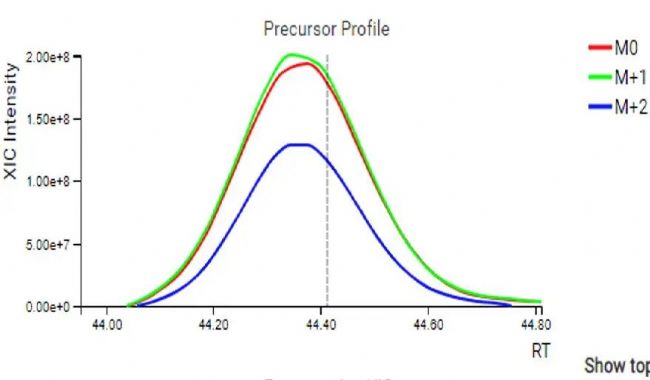
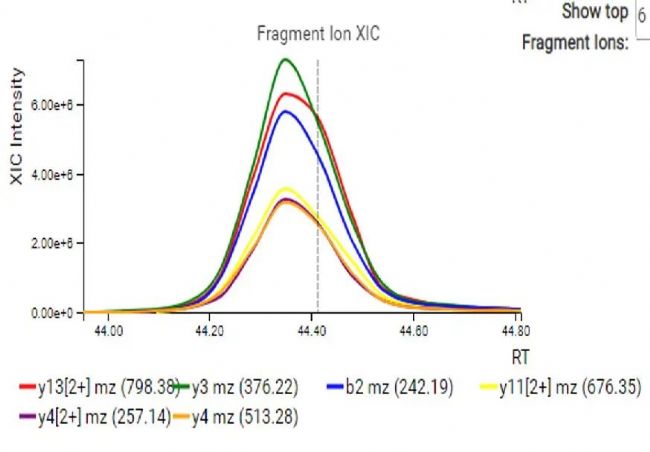
图5.母离子和碎片离子XIC
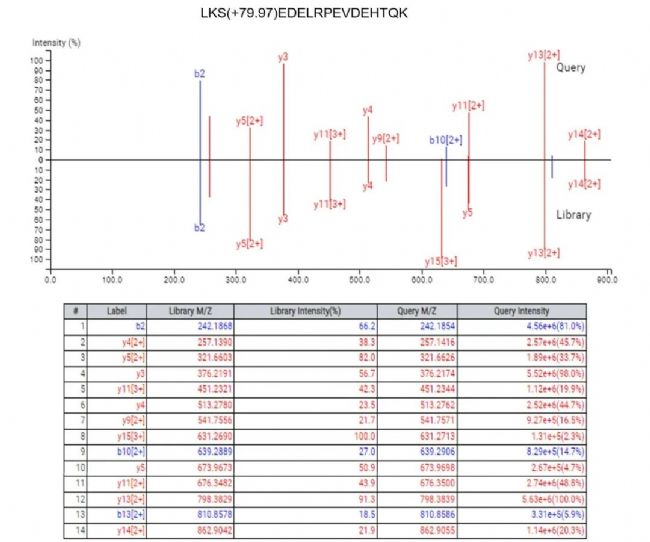
图6. 碎片离子对比与离子列表
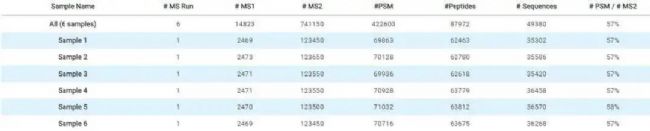
图7. 6个DIA数据的谱图库检索结果
PEAKS Online可以实现大规模数据集的快速分析。在PEAKS Online上通过DDA数据集构建谱图库,然后可以对复杂的DIA数据进行分析(Fig. 7)。
参考文献
2. Li, K.W., Gonzalez-Lozano, M.A., Koopmans, F. and Smit, A.B., 2020. Recent developments in data independent acquisition (DIA) mass spectrometry: application of quantitative analysis of the brain proteome. Frontiers in molecular neuroscience, p.248.

-扫码关注-
www.bioinfor.com (EN)
www.deepproteomics.cn(CN)
作为生物信息学的领军企业,BSI专注于蛋白质组学和生物药领域,通过机器学习和先进算法提供世界领先的质谱数据分析软件和蛋白质组学服务解决方案,以推进生物学研究和药物发现。我们通过基于AI的计算方案,为您提供对蛋白质组学、基因组学和医学的卓越洞见。旗下著名的PEAKS系列软件在全世界拥有数千家学术和工业用户,包括:PEAKS Studio,PEAKS Online,PEAKS AB及抗体综合表征服务等。
联系方式
021-60919891
sales-china@bioinfor.com



