
PEAKS DIA Streamlined工作流
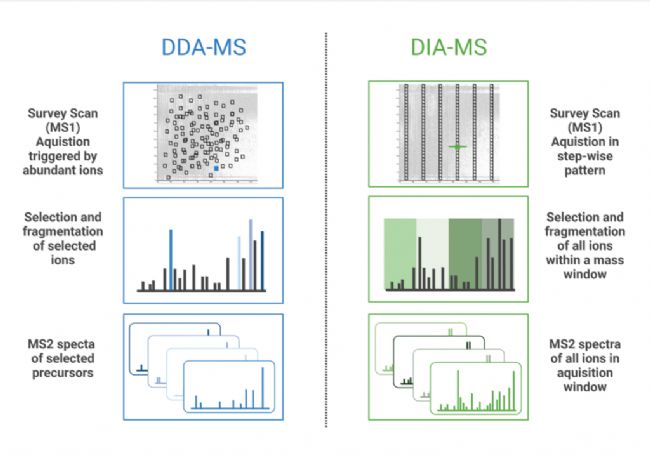
数据独立采集(DIA)是一种在设定的质荷比(m/z)窗口内的所有离子被碎裂和分析的质谱(MS)方法。与数据依赖采集(DDA)相反,DIA工作流可以不局限于丰度排名前几的母离子,而重复定性和定量多肽。在DDA模式下,质谱仪从MS1扫描中选择最高丰度的母离子,只有这些选择的肽段被串联质谱碎裂并分析。在DIA中,在给定时间内碎裂所有进入质谱仪的离子(broadband DIA)或在固定或可变的m/z窗口内依次碎裂离子 (SWATH,连续采集窗口获取所有理论碎裂离子),从而产生MS/MS 图谱(1)。近年的diaPASEF(平行累加-串联碎裂)技术,将捕集离子淌度分离(TIMS)与DIA相结合,以提高多肽检测的灵敏度并增加蛋白质组覆盖深度(2)。PEAKS Studio和Online都兼容上述所有类型的DIA数据,无论采用哪种方法,PEAKS软件都将帮助您的分析。
DIA工作流程的优点
- 减少分析的所有肽段的偏差
- 在Mass run中提高的多肽鉴定和定量的重复性(3)
- 在动态范围内量化复杂混合物中的蛋白质
- 消除抽样不足的影响
- 增加蛋白质组覆盖的灵敏度和深度
- 与DDA相比,精度和可重复性更高(4)
- 通过非标记定量降低成本
最大化提高DIA数据集的鉴定率
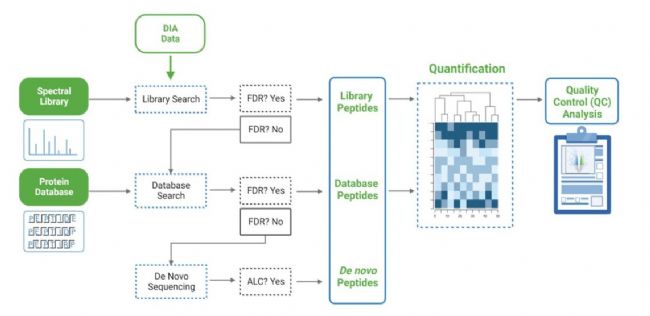
由于DIA MS/MS光谱是由一个m/z窗口内的一系列母离子产生的,它们通常包含来自多个多肽产生碎裂的信号。这对从DIA光谱中多肽鉴定算法提出了独特的挑战。PEAKS 11为解决这个问题提供了一个强大而灵活的解决方案,通过使用谱库搜索(无论是否包含用于蛋白质推断的序列数据库)、直接序列数据库搜索(direct Database)或几种方法的组合来进行肽定性和定量,以获得蛋白质组深度的肽段覆盖(5)。此外,可以应用从头测序来解析那些没有得到很好匹配的谱图。
对于谱图库和直接数据库搜索、从头测序这几种方式组合的分析,搜索空间是放大的。首先,对先前鉴定获得的谱图库进行SL search,通过错误发现率的阈值设定,保留通过筛选的多肽。在FDR阈值内没有得到良好匹配的谱图,进行直接数据库检索。来自与数据库匹配的可靠肽段会添加到结果中。然后,使用相同的FDR阈值设置,可以继续使用从头测序分析在数据库搜索中未匹配的谱图,即PEAKS Stream-lined工作流执行DIA的数据分析。
对于谱图库和直接数据库搜索、从头测序这几种方式组合的分析,搜索空间是放大的。首先,对先前鉴定获得的谱图库进行SL search,通过错误发现率的阈值设定,保留通过筛选的多肽。在FDR阈值内没有得到良好匹配的谱图,进行直接数据库检索。来自与数据库匹配的可靠肽段会添加到结果中。然后,使用相同的FDR阈值设置,可以继续使用从头测序分析在数据库搜索中未匹配的谱图,即PEAKS Stream-lined工作流执行DIA的数据分析。

DIA数据有几个可用的工作流程。传统上,常用的工作流程是从DDA数据生成一个库,并在搜索DIA数据时使用这个库,使得数据库搜索可以鉴定更多的多肽。另一种工作流程是使用PEAKS 11直接搜索DIA数据 (不需要事先搜索谱库),而且,未匹配的图谱可以进行从头测序,另外在定量模块中,可以对DIA数据进行非标记的定量。最后,在PEAKS Online中,QC分析提供了定性、定量的数据质控的统计概要和结果的可视化。
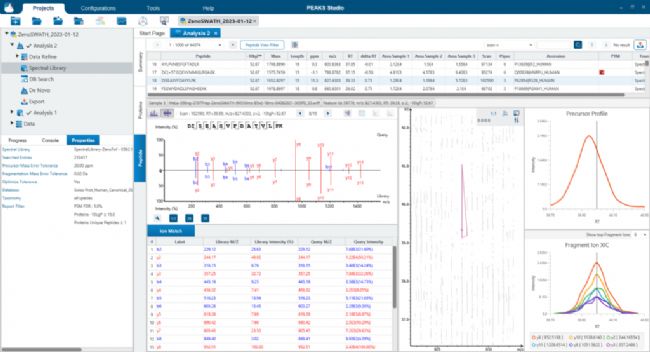 应用PEAKS SL search分析Sciex ZenoSWATH数据
应用PEAKS SL search分析Sciex ZenoSWATH数据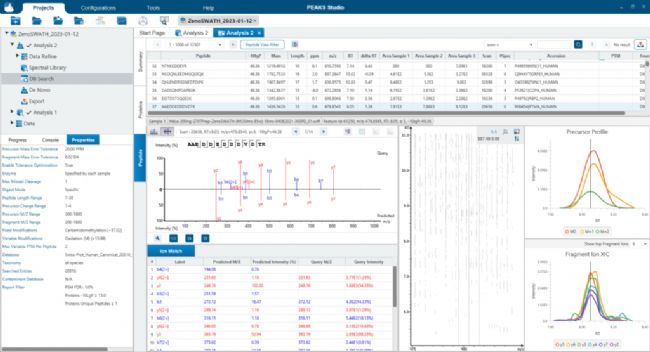 应用PEAKS Direct DB搜索分析Sciex ZenoSWATH数据
应用PEAKS Direct DB搜索分析Sciex ZenoSWATH数据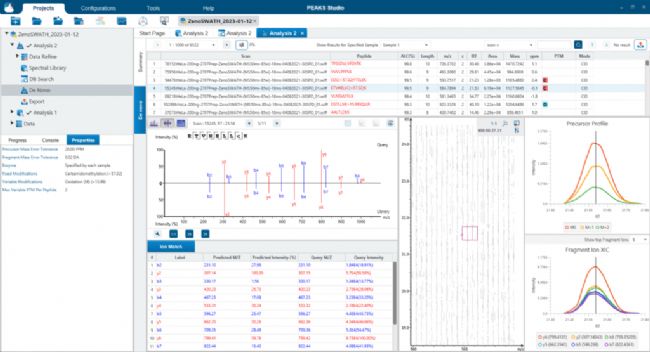 应用PEAKS DIA从头测序分析Sciex ZenoSWATH数据
应用PEAKS DIA从头测序分析Sciex ZenoSWATH数据定量具有更高的重现性
与DDA方法相比,DIA质谱产生高度可重复性的定量结果。谱图库和直接序列数据库搜索结果可以结合非标记定量方法,以帮助发现不同条件下的生物学显著变化。
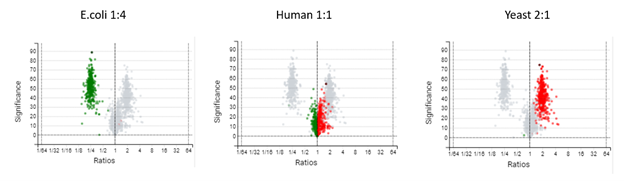 图1:人类、酵母和大肠杆菌混合样本的过滤火山图。根据不同的浓度,得到的比率符合预期。
图1:人类、酵母和大肠杆菌混合样本的过滤火山图。根据不同的浓度,得到的比率符合预期。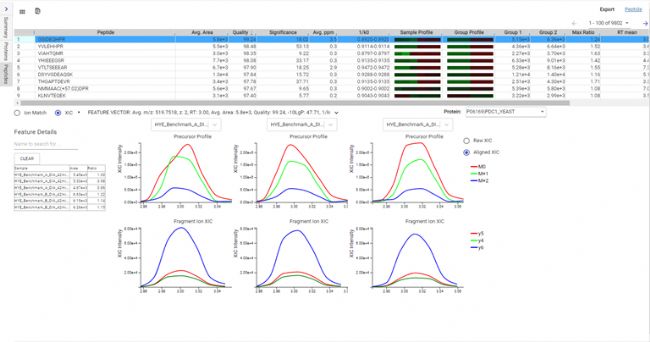
图2:非标记定量多肽母离子与碎片离子XIC曲线示例。最多可选择3个样本进行并排比较。
如果您想深入了解更多关于PEAKS DIA Streamlined 工作流,欢迎扫描下方二维码关注我们!




