

|
外显子测序(WES)是对具有蛋白编码功能的外显子进行的测序技术,近年来全外显子测序在发现外显子基因突变方面逐渐得到广泛应用。
|
|||||||||||||||||
| [发表评论] [本类其他服务] [本类其他服务商] | ||||||||||||||||||
| 服务商: 上海鲸舟基因科技有限公司 | 查看该公司所有服务 >> |
一、全外显子组测序(Whole Exome Sequencing,WES)
外显子测序(WES)是对具有蛋白编码功能的外显子进行的测序技术,近年来全外显子测序在发现外显子基因突变方面逐渐得到广泛应用。
外显子测序(WES)是对具有蛋白编码功能的外显子进行的测序技术,近年来全外显子测序在发现外显子基因突变方面逐渐得到广泛应用。
- 利用序列捕获技术将全基因组外显子区域DNA捕捉并富集后进行高通量测序。
- 外显子组测序的覆盖度更深,数据准确性更高;对于基因组重测序成本较低,对研究疾病相关的SNP、Indel等具有较大的优势。
- 基于大量的公共数据库提供的外显子数据,能够结合现有的资源更好地解释研究成果。
-
外显子组测序的优势
-
具有针对广泛应用领域的变异识别功能
-
实现编码区域的全面覆盖
-
提供一个可替代全基因组测序的高性价比方法(每个外显子组测序4–5 Gb,而每个全人类基因组测序约90 Gb)
-
与全基因组方法相比,生成的数据集更小、更易于管理,因此可以更简单快速地进行分析
有效地分析编码区域在不适合或不需要使用全基因组测序的情况下,外显子组测序不失为一种高性价比的方法。它只对基因组的编码区域进行测序,这样便将资源集中到最可能会影响表型的基因,因此能够一举为您缩短周转时间和节约成本。在检测编码外显子中的变异时,外显子组测序能够将靶向内容扩大到非翻译区(UTR)和microRNA,从而提供一个更全面的基因调控视野。仅需一天便能制备DNA文库,而且每个外显子组仅需测序4–5 Gb即可。
全外显子组测序实验流程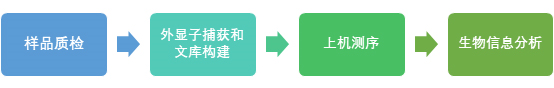
捕获平台:采用安捷伦最新的覆盖最全面的人外显子组捕获panel—SureSelect人全外显子V6
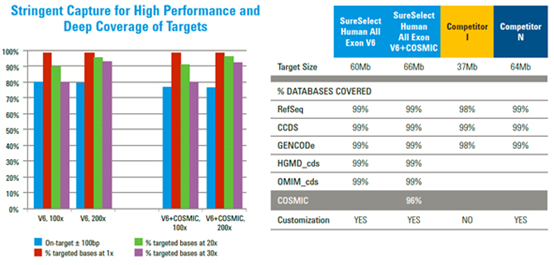
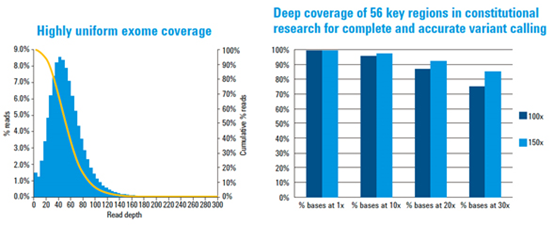
SureSelect 人全外显子 V6 高度优化的捕获探针设计结合严格的捕获工作流程,获得的高的在靶效率能够确保读出序列对靶标的特异性映射,从而实现深度覆盖。此外显子组靶向包括难捕获区域的相关数据库的更新内容,可实现蛋白质编码区域的全面分析。
样品要求² 样本类型DNA样品;样品浓度≥50ng/µl;样品总量≥2ug
数据分析
1. 标准分析:原始数据基本分析、序列比对、SNP检测等。
a) 基本信息分析:按Illumina 标准流程进行base calling、raw data 数据整理及数据质量评估;
b) 数据通过GATK, SAMtools 等检测SNP 和InDel 变异信息;
c) 将SNP 和InDel 与最新发布的dbSNP 和千人基因组数据进行比对分析;
d) 变异所在基因的功能注释;
e) 多样本分析。
2. 高级信息分析:孟德尔遗传疾病分析、复杂疾病分析、肿瘤癌症分析等。
全外显子组技术方案优势
1. 最全面的内容,适用于任何应用的外显子组解决方案- 包含相关数据库的最新核心内容,可靶向包括难捕获区域在内的更多外显子
- 轻松添加用于转化研究的 UTR、用于癌症研究的 COSMIC,或用于特定应用的定制内容
- 建议测序深度
- Germline变异相关建议测序深度80X以上(单基因/复杂疾病研究)
- Somatic变异相关建议测序深度150X以上(肿瘤癌症研究)
二、WES研究方案 — 基于全外显子组测序的孟德尔遗传疾病研究方案
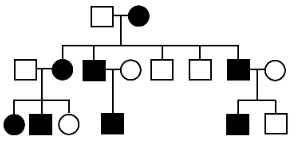
孟德尔遗传病,也称单基因遗传病,是指受一对等位基因(主效基因)控制的遗传性疾病。孟德尔遗传病是新生儿出生缺陷的重要原因之一,目前全球已知的单基因遗传病大约7000多种,而且其中大部分的潜在疾病基因还尚未研究清楚。
传统孟德尔疾病基因的识别主要是通过对候选基因实施Sanger测序而确定。候选基因的确定主要是通过一些基因组区域的位置映射方法,如核型分析,连锁分析等。然而,这种方法不能确定疾病是由某个单核苷酸突变导致还是基因组结构变异导致。随着二代测序技术的发展,尤其是外显子测序技术的出现,使得基因组以及基因组蛋白质编码区的常见及罕见变异都能被准确检测到,极大地促进疾病基因的识别。
研究目的
² 鉴定孟德尔遗传疾病中的致病位点
样本准备
² 家系样本:患病个体及核心家系成员(系谱图、临床表型、年龄)
² 测序深度建议:≥ 80X
² 遗传模式:常染色体显性遗传(AD);常染色体隐性遗传(AR);伴性遗传
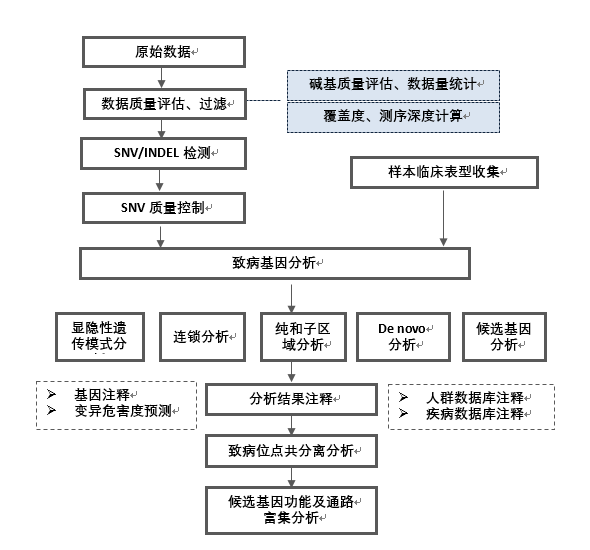
生物信息分析路线
三、WES研究方案 — 基于全外显子组测序的复杂疾病研究方案
由多个基因及环境因素相互作用所致的疾病,如心血管疾病、二型糖尿病、原发性高血压、银屑病等,称为复杂疾病(complex disease)或多基因病(polygenic disease)。这类疾病发病率一般都超过0.001,在临床或流行病学方面具有一定程度的家族倾向,但又不表现典型的孟德尔遗传方式。复杂疾病是一种受多基因、多因子影响的疾病,包括遗传因素、环境因素与其它因素。一般认为微效作用模式在复杂疾病的发生机制中起主要作用,即来自多个位点的大多数风险基因在群体中的发生频率都很低,它们之间有相互作用,通过数量性状的剂量效应关系,达到疾病发生的临界域值,而共同决定了复杂疾病的遗传易感性。
研究目的
² 探索复杂疾病的致病机制,寻找与疾病显著相关的致病和易感位点。
研究方法
进行复杂疾病的研究需要具备两个条件,一是要有遗传背景一致或相似的资源群体;二是要有覆盖全基因组的高密度的标记。利用和全外显子组测序,我们可在全外显子组范围内获取大量的准确性高的变异位点。
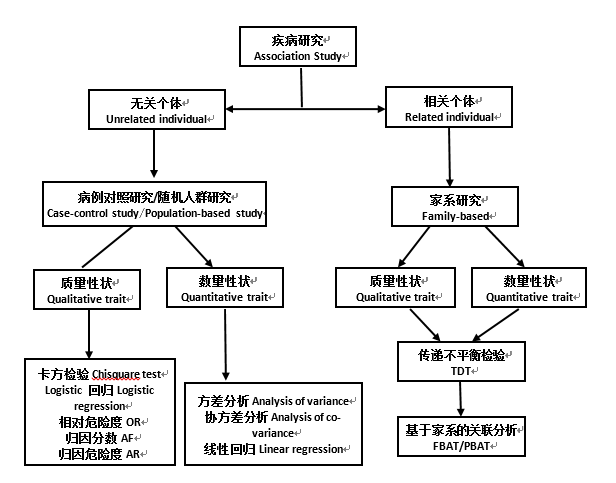
复杂疾病遗传变异的研究依据研究对象不同的遗传背景来源,分为基于家系和基于散发人群两种研究策略。目前常规的分析方法如下图所示:
 样本准备
样本准备
- Case-control散发样本:疾病患者与正常对照样本的外周血DNA。Case样本数建议>200; control样本数建议>200。样本来源保持地域一致,例如:少数名族与汉族的分开。疾病分组若 按亚型来分,每种 亚型建议样本数>100。
- 家系样本:患病个体及核心家系成员(系谱图、个体表型、年龄)。
- 测序深度建议:≥ 80X。
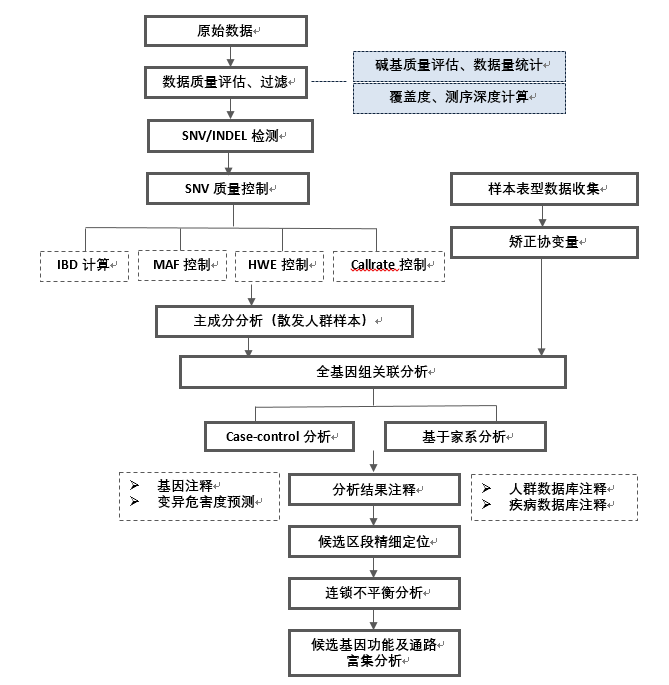
生物信息分析路线
四、WES研究方案 —基于全外显子组测序的肿瘤驱动基因(driver mutation)研究方案
为什么会产生癌症?我们需要提到一个名词:驱动基因,它可以说是决定癌症的最主要原因。如何找到驱动基因,是制定癌症治疗措施的关键所在。根据目前的发现,驱动基因包括:突变的EGFR、ALK融合基因、突变的KRAS、突变的HER2等的一大类都是驱动基因。例如,肺癌可按照驱动基因分为许多类,这是近年来非常重要的一个发现。由此可以根据不同的驱动基因,采用合适的药物进行治疗。如果找到驱动基因,治疗就事半功倍,如果没有找到驱动基因而盲目治疗,靶向治疗效果较差,可能连安慰剂都不如。
全外显子组测序是用于分析基因组蛋白质编码区域的一种功能强大的技术。这种大规模并行式分析结合了新一代测序的单分子分辨率,凭借能够分析约含 20,000 个基因的外显子基因组的高性价比单反应方法,已经在加速疾病基因发现中发挥了重要作用。
研究目的
² 利用配对的肿瘤样本进行高通量测序,检测导致肿瘤发生的Driver mutation。
样本准备 - 血液样本:EDTA抗凝,2ml,-20℃冰箱保存。
- 新鲜冰冻组织:液氮冷却、-80℃冰箱保存。
- FFPE样本:FFPE切片(10片左右,切片厚度在10μm左右)
测序深度建议 - 肿瘤样本 ≥ 150X
- 血液及正常组织样本≥ 100X
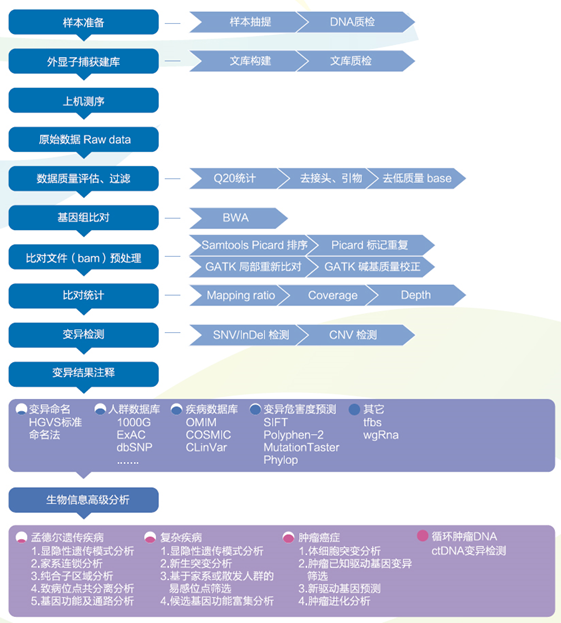
五、全外显子组测序技术路线与生物信息分析展示
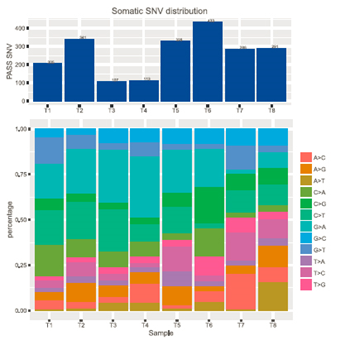
1.突变频率分布图
2. 突变功能分布图

图中展示了其中一个样本突变功能分布情况。上方的饼图表示在基因不同区域上(intron, exon, etc.)的变异所占的百分比;下方的饼图表示在exon位置上不同变异类型所占的百分比。
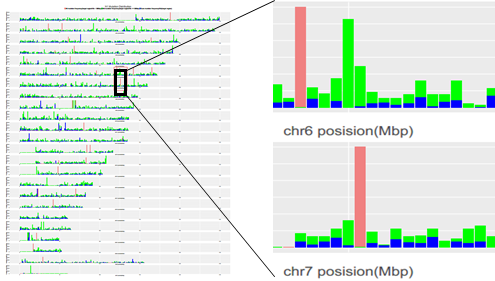
3.样本拷贝数变异区间图
图形上方展示每对样本体细胞突变数目,下图展示每对样本体细胞突变不同突变类型所占的比例。
5. 样本体细胞突变heatmap图
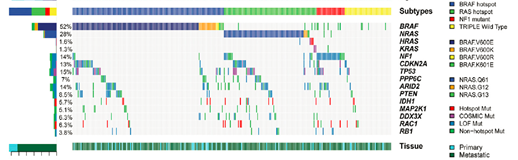
6. 体细胞突变富集分析图

左边纵坐标表示Go term,横坐标是富集因子(Rich Factor = Gene number/Total Gene Number of the term)。每个圆圈的大小与在这个Go Term上的基因成正比,颜色与根据q-value的log值从红到绿渐变,越红,q-value值越低,富集越显著。
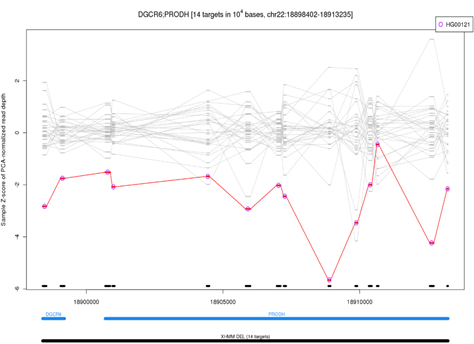
7. 家系连锁分析LOD连锁图及单倍型图
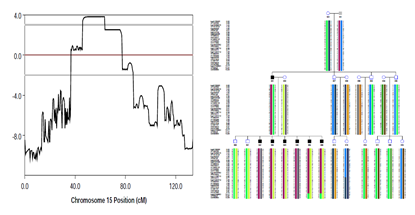
左图为LOD连锁图,通常,LOD值≥3被认为是两个基因连锁得确定证据(因为LOD值是以10为底的对数计算的,LOD值为3表明1000:1的机会,即所观察到的连锁不是偶然发生的);LOD值≥2时,可能连锁;-2<LOD值<2时,需要更多地家系资料,尚不能判别是否连锁;LOD值<-2时,为不连锁;右图为致病位点单倍型图,可以根据此图判断致病位点是否在家系患病成员中处于共分离状态。
8. 曼哈顿图(Manhattan Plot)
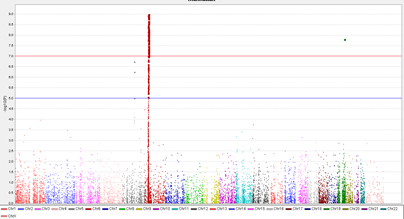
X-轴为基因组坐标,Y-轴为每个单核苷酸多态性的关联P-值的负对数。
相关解决方案
癌症全外显子组测序 癌症全外显子组测序提供了关于导致肿瘤恶化的编码区突变的有用信息。通过测序全基因组的2%区域,能够更为经济地获得更高的测序深度。了解癌症外显子组测序的更多信息。致病变异的发现
癌症全外显子组测序提供了关于导致肿瘤恶化的编码区突变的有用信息。通过测序全基因组的2%区域,能够更为经济地获得更高的测序深度。了解癌症外显子组测序的更多信息。致病变异的发现 对单个和多个个体的全外显子组测序是寻找罕见病、复杂病或孟德尔病致病变异的常用方法。遗传关联与连锁分析经由人群中普遍的遗传变异,提供了复杂疾病研究的基础。了解致病变异发现的更多信息。转化研究
对单个和多个个体的全外显子组测序是寻找罕见病、复杂病或孟德尔病致病变异的常用方法。遗传关联与连锁分析经由人群中普遍的遗传变异,提供了复杂疾病研究的基础。了解致病变异发现的更多信息。转化研究 外显子组测序正越来越多地应用于转化和临床研究以了解基因在各种人类疾病中所扮演的角色。 了解转化研究的更多信息。
外显子组测序正越来越多地应用于转化和临床研究以了解基因在各种人类疾病中所扮演的角色。 了解转化研究的更多信息。
手机版:全外显子组测序服务

Copyright(C) 1998-2025 生物器材网 电话:021-64166852;13621656896 E-mail:info@bio-equip.com