
戊二酸血症模型Gcdh KO小鼠在罕见代谢疾病研究中的应用
戊二酸血症I型(GA1),又称戊二酸尿症I型,是一种常染色体隐性遗传的代谢性疾病。2018年,该疾病被列入国家卫健委等五部门联合制定的《第一批罕见病目录》。GA1由L-赖氨酸、L-羟基赖氨酸和L-色氨酸的分解代谢缺陷引起,导致代谢产物戊二酸(GA)、3-羟基戊二酸(3-OH-GA)及戊二酰肉碱(C5DC)在体内异常蓄积,进而造成代谢紊乱,主要影响神经系统。GA1的全球发病率约为1/100,000(儿童发病率约为1/30,000至1/100,000)[1],具有种族和地域差异,国内报道约为1/60,000。GA1患儿在正常发育阶段后,可能因感染、疫苗接种或手术等诱因触发急性脑病,导致不可逆的纹状体损伤,具有较高的致死致残率。
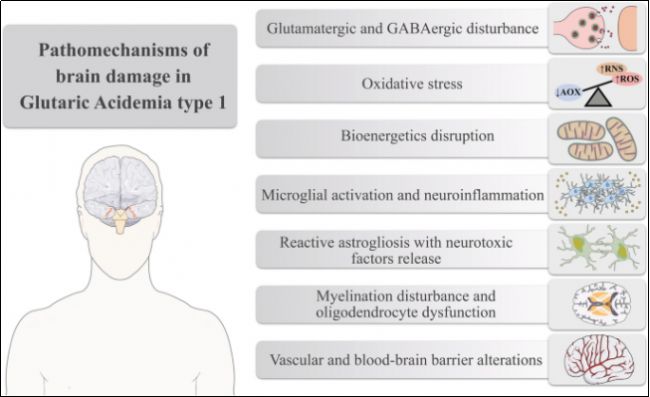
图1 GA1疾病中脑部损伤的机制 [2]
1.戊二酸血症I型的发病机制
戊二酰辅酶A脱氢酶(GCDH)是一种线粒体酶,属于脱氢酶/脱羧酶酶类家族,主要分布于肝脏、肾脏及脑部等代谢活跃组织的线粒体中。GCDH的主要功能是催化戊二酰辅酶A(Glutaryl-CoA, GA-CoA)氧化生成戊烯二酰辅酶A(Glutaconyl-CoA),随后进一步脱羧为巴豆酰辅酶A(Crotonyl-CoA)。这一过程是赖氨酸、羟基赖氨酸及色氨酸分解代谢的关键步骤。由于这些氨基酸是必需氨基酸,其代谢路径中的中间产物需及时清除,避免在体内蓄积造成毒性。此外,GCDH的功能缺陷会导致某些代谢途径中断,影响能量供给,对大脑等高度依赖能量组织的影响尤为明显。在GCDH缺乏的情况下,戊二酰辅酶A无法正常代谢,导致其转化为戊二酸(GA)、3-羟基戊二酸(3-OH-GA)及戊二酰肉碱(C5DC)等毒性代谢物。这些代谢物对中枢神经系统具有高度毒性,特别是在纹状体区域,可能引发神经元损伤、神经元空泡化及炎症反应 [3-5]。临床表现包括巨脑症、进行性肌张力障碍和运动障碍,严重者可能致命。
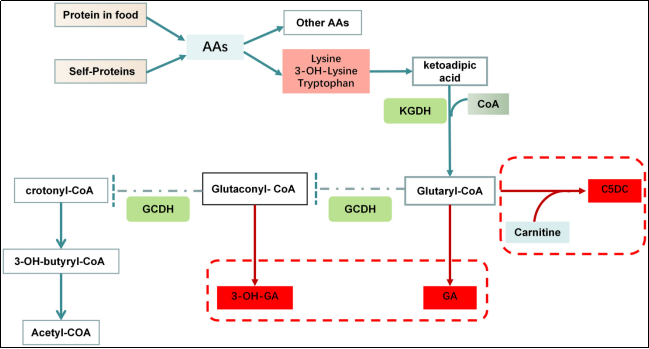
图2 GA1中的赖氨酸和色氨酸代谢紊乱 [5]
2.Gcdh KO小鼠模型的研究价值
研究表明,Gcdh基因敲除小鼠(Gcdh KO小鼠)展现出与人类GA1疾病高度相似的生化表型,其尿液和脑组织中GA及3-OH-GA水平显著升高,血清中C5DC含量亦显著增加,与患者体内水平一致。此外,Gcdh KO小鼠在高蛋白饮食条件下会出现脑病风险、脑空泡形成,并于4~5天内死亡 [7]。高赖氨酸饮食(HLD)进一步加重表型,表现为代谢物积累、纹状体神经变性和年龄相关脑损伤,断奶时喂食HLD的小鼠致死率显著升高。存活至成年期的KO小鼠常伴有严重的神经病理学改变,包括神经元缺失、空泡化及脑室内出血 [8-9]。因此,Gcdh KO小鼠被广泛用于GA1疾病机制研究、治疗药物开发及药效评估 [10-13]。
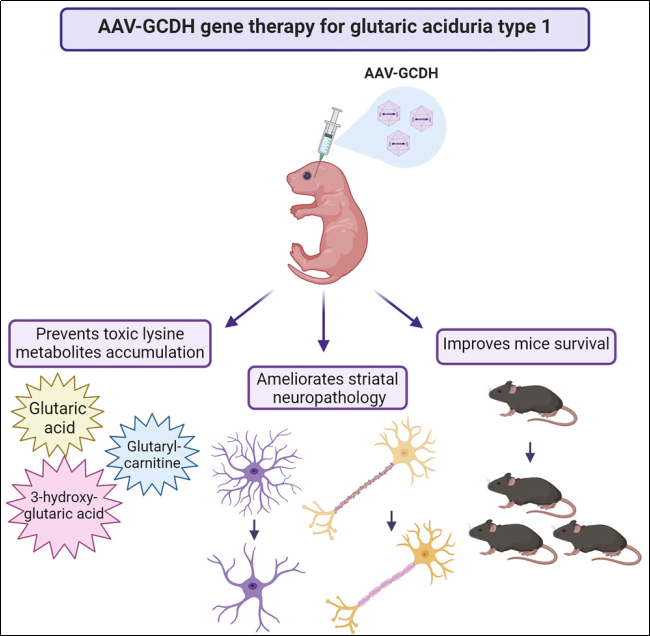
图3 Gcdh KO小鼠用于腺相关病毒(AAV)介导补充疗法的临床前药效评估 [10]
3.赛业生物Gcdh KO小鼠模型
赛业生物通过敲除Gcdh基因构建了Gcdh KO小鼠(产品编号:C001594)。该模型在血浆、脑部和肝脏组织中均累积了大量的戊二酸(GA),与野生型小鼠相比,呈现出典型的戊二酸血症I型(GA1)疾病生化表型。该模型是研究GA1发病机制的理想工具,可用于药物研发、氨基酸代谢及GCDH功能研究。
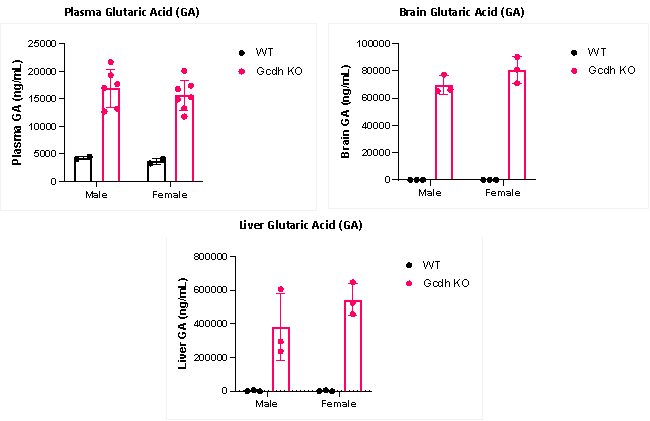
图4 野生型小鼠(WT)与Gcdh KO小鼠体内GA水平对比
参考文献
[1]Li Q, Yang C, Feng L, Zhao Y, Su Y, Liu H, Men H, Huang Y, Körner H, Wang X. Glutaric Acidemia, Pathogenesis and Nutritional Therapy. Front Nutr. 2021 Dec 15;8:704984.
[2]Wajner, M. (2022). Glutaric Acidemia Type 1: An Inherited Neurometabolic Disorder of Intoxication. In: Kostrzewa, R.M. (eds) Handbook of Neurotoxicity. Springer, Cham.
[3]Schuurmans IME, Dimitrov B, Schröter J, Ribes A, de la Fuente RP, Zamora B, van Karnebeek CDM, Kölker S, Garanto A. Exploring genotype-phenotype correlations in glutaric aciduria type 1. J Inherit Metab Dis. 2023 May;46(3):371-390.
[4]Boy N, Mühlhausen C, Maier EM, Ballhausen D, Baumgartner MR, Beblo S, Burgard P, Chapman KA, Dobbelaere D, Heringer-Seifert J, Fleissner S, Grohmann-Held K, Hahn G, Harting I, Hoffmann GF, Jochum F, Karall D, Konstantopoulous V, Krawinkel MB, Lindner M, Märtner EMC, Nuoffer JM, Okun JG, Plecko B, Posset R, Sahm K, Scholl-Bürgi S, Thimm E, Walter M, Williams M, Vom Dahl S, Ziagaki A, Zschocke J, Kölker S. Recommendations for diagnosing and managing individuals with glutaric aciduria type 1: Third revision. J Inherit Metab Dis. 2023 May;46(3):482-519.
[5]Li Q, Yang C, Feng L, Zhao Y, Su Y, Liu H, Men H, Huang Y, Körner H, Wang X. Glutaric Acidemia, Pathogenesis and Nutritional Therapy. Front Nutr. 2021 Dec 15;8:704984.
[6]Koeller DM, Woontner M, Crnic LS, Kleinschmidt-DeMasters B, Stephens J, Hunt EL, Goodman SI. Biochemical, pathologic and behavioral analysis of a mouse model of glutaric acidemia type I. Hum Mol Genet. 2002 Feb 15;11(4):347-57.
[7]Keyser B, Glatzel M, Stellmer F, Kortmann B, Lukacs Z, Kölker S, Sauer SW, Muschol N, Herdering W, Thiem J, Goodman SI, Koeller DM, Ullrich K, Braulke T, Mühlhausen C. Transport and distribution of 3-hydroxyglutaric acid before and during induced encephalopathic crises in a mouse model of glutaric aciduria type 1. Biochim Biophys Acta. 2008 Jun;1782(6):385-90.
[8]Zinnanti WJ, Lazovic J, Wolpert EB, Antonetti DA, Smith MB, Connor JR, Woontner M, Goodman SI, Cheng KC. A diet-induced mouse model for glutaric aciduria type I. Brain. 2006 Apr;129(Pt 4):899-910.
[9]Seminotti B, Amaral AU, da Rosa MS, Fernandes CG, Leipnitz G, Olivera-Bravo S, Barbeito L, Ribeiro CA, de Souza DO, Woontner M, Goodman SI, Koeller DM, Wajner M. Disruption of brain redox homeostasis in glutaryl-CoA dehydrogenase deficient mice treated with high dietary lysine supplementation. Mol Genet Metab. 2013 Jan;108(1):30-9.
[10]Mateu-Bosch A, Segur-Bailach E, Muñoz-Moreno E, Barallobre MJ, Arbonés ML, Gea-Sorlí S, Tort F, Ribes A, García-Villoria J, Fillat C. Systemic delivery of AAV-GCDH ameliorates HLD-induced phenotype in a glutaric aciduria type I mouse model. Mol Ther Methods Clin Dev. 2024 Jun 4;32(3):101276.
[11]Barzi M, Johnson CG, Chen T, Rodriguiz RM, Hemmingsen M, Gonzalez TJ, Rosales A, Beasley J, Peck CK, Ma Y, Stiles AR, Wood TC, Maeso-Diaz R, Diehl AM, Young SP, Everitt JI, Wetsel WC, Lagor WR, Bissig-Choisat B, Asokan A, El-Gharbawy A, Bissig KD. Rescue of glutaric aciduria type I in mice by liver-directed therapies. Sci Transl Med. 2023 Apr 19;15(692):eadf4086.
[12]Wagner GR, Bhatt DP, O'Connell TM, Thompson JW, Dubois LG, Backos DS, Yang H, Mitchell GA, Ilkayeva OR, Stevens RD, Grimsrud PA, Hirschey MD. A Class of Reactive Acyl-CoA Species Reveals the Non-enzymatic Origins of Protein Acylation. Cell Metab. 2017 Apr 4;25(4):823-837.e8.
[13]Sauer SW, Opp S, Komatsuzaki S, Blank AE, Mittelbronn M, Burgard P, Koeller DM, Okun JG, Kölker S. Multifactorial modulation of susceptibility to l-lysine in an animal model of glutaric aciduria type I. Biochim Biophys Acta. 2015 May;1852(5):768-77.



