
CNVkit工具详解:既简又快地检测外显子拷贝数变异
人类染色体在遗传过程中,有时候会丢失或增加一部分,遗传学将这种DNA的增减现象称为拷贝数变异(CNV)。CNV通常为长度大于1kb的片段变异,里面往往包含一个或多个基因,若变异片段包含功能特别重要、对剂量水平敏感的基因,就会导致疾病的发生,可导致智力低下、发育异常等症状,引起残疾、甚至死亡。
外显子不同于基因组,由于是捕获测序,本身就是各个外显子区域分割开来,这样造成2个影响:(1)大片段的CNV被切开形成多个假阳性;(2)只能检测小的CNV。因此对检测软件要求很灵敏。
目前有很多检测CNV的工具,比如CNVseq、CNVnator/CNVpytor、Conifer、WisecondorX等,但这些工具要么是安装难度大,要么是不适合外显子CNV检测,又或者使用繁琐。有没有一种安装方便、用法简单、适合外显子CNV检测的工具?就是今天要说的CNVkit。
CNVkit于2016年发表在PLOS computational biology上,详见《CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing》。CNVkit是基于Python3.7+开发的一款专门用于基因组高通量测序数据拷贝数变异(CNV)检测工具,并提供多种可视化方法。
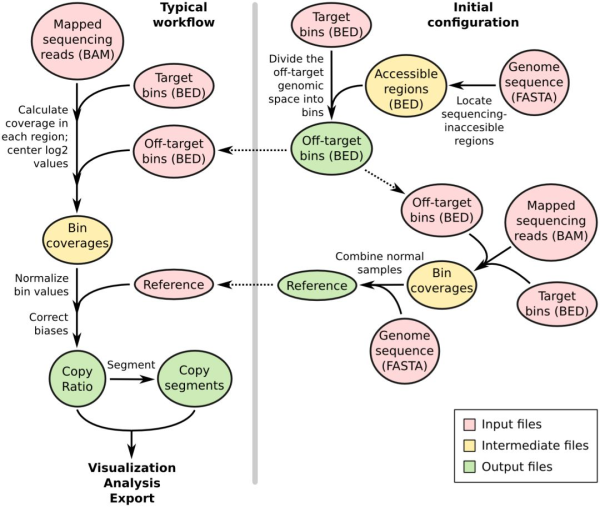
CNVkit分析流程示意图
该方法将in-target和off-target区域划分成小的bin区间,统计每个bin区间内的测序深度、GC含量,并通过GC含量、目的区域的大小和分布密度等对原始测序深度进行校正,去除肿瘤样本中的背景噪音,然后计算肿瘤样本相对正常样本对应位置拷贝数的log2 ratio值, 最后采用segment算法进行过滤,得到最终的肿瘤样本拷贝数变异结果。
软件安装使用介绍
1、通常我们使用conda安装,另外需要额外安装R包DNAcopy,因为CNVkit内部需要借助DNAcopy包进行拷贝数估算,安装没什么幺蛾子,比较顺利。
2、需要下载基因注释文件,用于对CNV结果进行基因注释,这倒省了我们单独注释的麻烦。该文件可以在UCSC上下载
PS:如果需要做hg38或其他物种,可以从NCBI下载物种基因组注释文件(gtf或gff格式),提取信息自行制作refFlat.txt.gz文件,格式跟UCSC下载的保持一致即可。
3、参考基因可在NCBI或UCSC上下载,需要与refFlat.txt.gz文件基因信息一致。
4、样本bam数据,参考BWA流程获得。
5、一键运行:
$cnvkit access $ref -s $window -o $out/access-${window}.bed
$cnvkit.py batch --method hybrid $Tumor.bam \
--normal $Normal.bam\
--annotate $refFlat \
--fasta $ref \
-t $target.bed \
--access $out/access-${window}.bed \
-d $output \
--segment-method hmm \
--diagram \
--scatter \
-p 6
PS:使用CNVkit的batch命令,可以一步完成所有分析步骤,最后用--diagram和--scatter用于绘制拷贝数变异总览图和染色体分布图,不需要可以不使用。
文献摘录
这里介绍一篇发表于2022年的文章《Gliosarcoma: The Distinct Genomic Alterations Identified by Comprehensive Analysis of Copy Number Variations》,研究者使用CNVkit对胶质母细胞瘤(GBM)和胶质肉瘤(GSM)的DNA拷贝数变异(CNV)进行了全面分析,下图就展示了21例GBM 和15例GSM 样本拷贝数变异在染色体上的分布(该图使用CNVkit自带参数heatmap生成)。
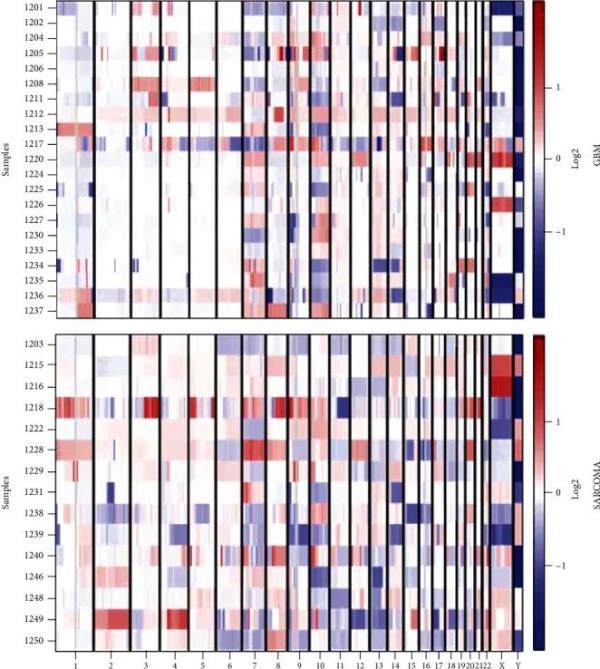
Figure 1 横坐标是染色体,纵坐标是样本,红色表示拷贝数扩增,蓝色表示拷贝数缺失,颜色越深,表示拷贝数扩增/缺失越多



