
高分文章解读:铁死亡在肿瘤,造血干细胞,心血管疾病中的调控机制
8月24日,一年一度的国家自然科学基金又重磅放榜啦。2023年度受理期间,国家自然科学基金委员会共接收项目申请超过30万项,其中有2万项通过审批获得了项目资助。查阅相关的中标项目,我们不难发现一些眼熟的关键词仍然备受青睐:
| 巨噬细胞 | 干细胞 |
| 线粒体 | 甲基化 |
| 炎症 | 细胞衰老 |
| 自噬 | 细胞焦亡 |
| 铁死亡 | 肿瘤微环境 |
铁死亡作为一种新型的细胞死亡机制相信大家已经不陌生,近几年来铁死亡的热度一直居高不下,2023年更是有超过500项标书立项!除了连续称霸国自然热度榜以外,铁死亡也是近几年高分杂志的宠儿,频频摘得热门杂志的青睐。这么火爆的热点大家还不追嘛?今天小优就用几篇近3年的高分文献带大家深度了解铁死亡的研究方向和方法。因为篇幅有限,这次我们先分享其中的5篇文章,剩下的文章分享请继续关注我们下篇文章。
铁死亡是一种铁依赖性的,非细胞凋亡性的细胞死亡形式,主要是细胞内脂质活性氧(ROS)生成与降解的平衡失调所致。对铁死亡还不了解的小伙伴可以点击查看我们之前的文章分享:53分铁死亡相关Review,究竟写了啥?近几年铁死亡的研究已经深入到各个系统和疾病,其中在癌症研究,肿瘤研究,免疫疗法等领域都有着非常热门的研究。话不多说,接下来我们就一睹为快吧!
1、Ferroptosis surveillance independent of GPX4 and differentially regulated by sex hormones
发表期刊:Cell(IF:64.5)
发表时间:2023年6月
铁死亡是一个铁依赖性的磷脂过氧化反应诱导的细胞死亡程序,已知有两个主要的监察机制来抑制铁死亡:分别是glutathione peroxidase 4 (GPX4)和其他的介导的酶例如FSP1,DHODH,NOS2和GCH1。除了现存的GPX4和RTAs通路以外是否还有其他的机制?来自纪念斯隆-凯特琳癌症中心的姜学军团队对此问题进行探究,揭示了一种受性激素调控的新监测机制。
在本篇文章中,作者通过全基因组 CRISPR 激活筛选,然后进行机理研究,发现磷脂修饰酶 MBOAT1 和 MBOAT2 是铁死亡的抑制因子。MBOAT1/2通过重塑细胞磷脂结构来抑制铁突变,他们的铁死亡监视功能不依赖于GPX4或FSP1。MBOAT1 和 MBOAT2通过性激素受体(即雌激素受体(ER)和雄激素受体(AR))上调。将ER或AR拮抗剂与铁死亡诱导相结合,可显著抑制ER+乳腺癌和AR+前列腺癌。
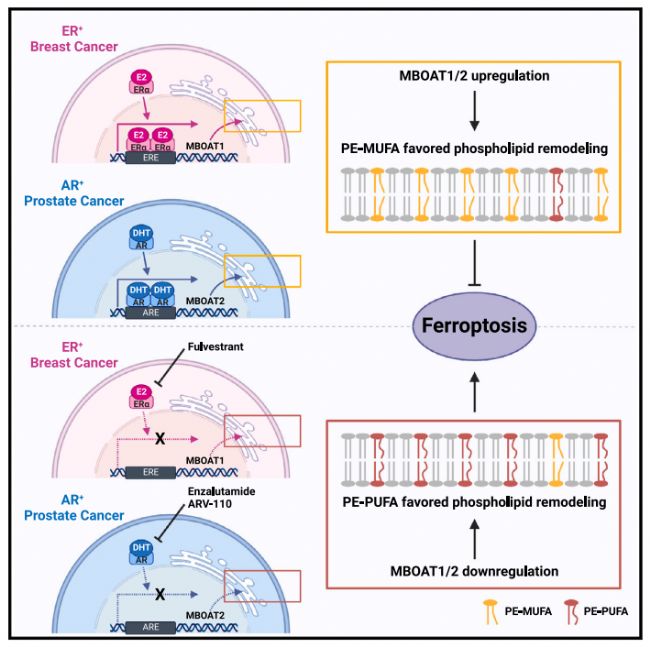
文章的大概思路如下:
1.为了鉴定铁死亡抑制的基因,作者用GPX4抑制剂RSL3和铁死亡诱导物(FINs)在人纤维瘤细胞HT1080细胞中构建了全基因CRISPR激活筛选系统,作者发现了一个新的铁死亡抑制基因MBOAT2。经过验证,MBOAT2是一个铁死亡抑制基因,不依赖于GPX4和FSP1。
2.作者接下来验证MBOAT2抑制铁死亡是否通过磷脂(PL)的新陈代谢。结果显示MBOAT2介导的铁死亡活性抑制依赖于内源性和外源性的单不饱和脂肪酸MUFA。
3.由于MBOAT2抑制铁死亡是一个MUFA依赖性的行为,接下来作者继续验证MBOAT2抑制铁死亡是否通过PL重塑。结果表明MBOAT2抑制铁死亡通过磷脂重塑,MBOAT2在对抗内源性的MUFA诱导的铁死亡中起到重要作用。
4.通过对比多种正常组织和癌症组织的MBOAT2表达水平,作者发现MBOAT2在前列腺癌症(PCa)中特异性上调。进一步研究发现,雄激素受体(AR)信号通路调节铁死亡依赖于MBOAT2。并且AR信号通路促进细胞对铁死亡的耐受通过上调MBOAT2的表达和重塑细胞PL。
5.因为在AR+ PCa中上调MBOAT2可以促进铁死亡耐受,作者继续研究AR拮抗剂是否可以作为一个治疗策略通过下调MBOAT2使肿瘤细胞对铁死亡更敏感。结果表明AR拮抗剂使AR+前列腺癌症对铁死亡敏感。
6.作者对MBOAT家族进行进一步研究,发现MBOAT1也可以抑制铁死亡。AR可以调节MBOAT2而不调节MBOAT1,而ER可以调节MBOAT1而不调节MBOAT2。结果表明,MBOAT1和MBOAT2通过不同途径调节铁死亡。
7.作者接下来继续研究是否ER和AR拮抗剂可以激活ER+ BCa对铁死亡的敏感性通过下调MBOAT。结果表明将ER或AR拮抗剂与铁死亡诱导相结合,可显著抑制ER+乳腺癌和AR+前列腺癌。
综上所述,这一系统性的研究揭示了一种不依赖于GPX4或FSP1的新的调控机制,性激素信号通过MBOAT1/2介导的PL重塑抑制癌症细胞铁死亡,为癌症治疗提供新的思路。
文章中使用到的技术主要有:构建CRISPR敲除小鼠,CRISPR激活检测和数据分析,RNA测序,ChIP,构建耐药小鼠模型
2、Human hematopoietic stem cell vulnerability to ferroptosis
发表期刊:Cell(IF:64.5)
发表时间:2023年2月
造血干细胞(HSCs)具有许多独特的机制来维持血液细胞的生成,来自丹娜法伯癌症研究所的Vijay G. Sankaran团队发现人类造血干细胞容易收到铁死亡的影响,并对其机制做了深入的研究。
作者从一种因组蛋白去泛素化酶 MYSM1缺失导致的骨髓衰竭疾病得到启发,表明造血干细胞蛋白质合成减少如何导致铁死亡增加。尽管蛋白质生成速率没有改变,通过阻断铁死亡可以完全挽救造血干细胞的功能。重要的是,这种对铁死亡选择性的脆弱不仅是MYSM1缺乏时造血干细胞丧失的基础,也阐释了人类造血干细胞广泛的依赖性。通过过表达MYSM1提高蛋白质合成速率,可降低造血干细胞对铁死亡的敏感性。
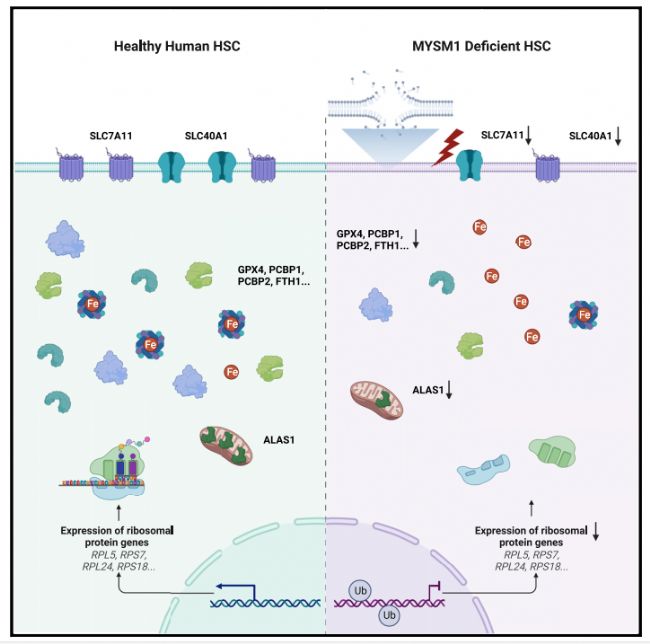
文章的大概思路如下:
1.骨髓衰竭综合征是由于MYSM1的突变引起的,作者想探究MYSM1和造血干细胞之间是否有关联。经过实验表明,MYSM1缺失会损伤人HSC功能。
2.接下来作者验证MYSM1如何影响HSC的功能。作者通过RNA测序发现和核糖体相关的基因都有明显下调。在MYSM1缺失的HSC中,核糖体蛋白水平明显下降。作者又进一步检测了蛋白生成率,发现MYSM1缺失引起HSCs蛋白生成率下降。
3.接下来作者继续研究MYSM1如何影响HSC功能。作者发现在MYSM1缺失的HSC中,氧化剂解毒,抗氧化剂,和铁结合基因明显上调,有关铁新陈代谢和抗氧化相关的基因上调,经过进一步研究发现,MYSM1缺失引起HSC功能影响是因为铁死亡。而抑制铁死亡可以缓解MYSM1缺失带来的HSC功能影响。
4、作者接下来进一步研究铁死亡和HSC功能失调相关的具体机制。作者检测了铁死亡相关的基因和通路,结果发现在MYSM1敲除的细胞中保护细胞免受铁死亡的基因都有明显的下调。MYSM1缺失造成的转录速率下调是通过铁死亡保护的基因转录效率下降而影响HSC功能。
综上所述:这一系统性的研究揭示了MYSM1缺失通过铁死亡导致HSC功能影响,此研究为治疗药物耐受的癌症和棘手的血液疾病提供新的临床思路。
文章中使用到的技术主要有:构建转基因小鼠模型,CRISPR/Cas9,RNA测序,流式,电镜检测
3、A non-canonical vitamin K cycle is a potent ferroptosis suppressor
发表期刊:Nature(IF:64.8)
发表时间:2022年8月
铁死亡是一种非凋亡性细胞死亡形式,以铁依赖性脂质过氧化为特征,在器官损伤、退行性疾病和耐药性癌症的易感性中起着关键作用。来自德国亥姆霍兹慕尼黑研究中心的 Marcus Conrad 团队发现了维生素K在介导铁死亡中新功能,完全还原形式的维生素K可作为一种抗氧化剂,有效抑制细胞铁死亡。
在文章中作者展示了维生素 K 的完全还原形式——萘醌(包括甲萘醌和植物醌)除了作为γ-谷氨酰羧化酶的辅助因子与血液凝固有关的常规功能外,还具有很强的抗铁死亡功能。研究发现,铁死亡抑制蛋白1(FSP1)是一种 NAD(P)H-泛醌还原酶,它能有效地将维生素 K 还原成氢醌,而氢醌是一种强效的自由基捕获抗氧化剂和(磷)脂质过氧化抑制剂。另外,FSP1 介导的维生素 K 还原对warfarin中毒也有解毒作用。由此可见,FSP1 是介导warfarin耐受的维生素 K 还原的酶。 FSP1 依赖的非经典维生素 K 循环可保护细胞免受有害的脂质过氧化和铁死亡。
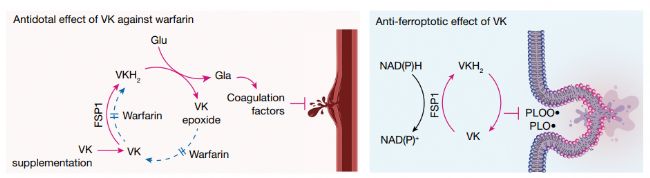
文章的大概思路如下:
1.为了探究是否还有其他机制能有效抑制铁死亡,作者在他莫昔芬(TAM)诱导Gpx4缺失的小鼠胚胎成纤维细胞(简称Pfa1细胞)中系统地研究了多种天然维生素化合物。除了α-生育酚(α-TOH)这种生物活性最强的维生素E外,作者发现有三种形式的维生素K——即植物醌、甲萘醌-4(MK4)和甲萘醌,能使细胞免遭Gpx4缺失诱导的铁死亡。进一步研究发现,这三种形式的维生素K都能有效抑制脂质过氧化。
2.接下来作者继续检测维生素K是否也能在体内抑制铁死亡。作者将TAM诱导的肝细胞特异性Gpx4缺失小鼠中,在维生素E缺乏的条件下,超营养水平的甲萘醌-4(MK4)延长了小鼠的存活时间,并有力保护了其肝脏。在小鼠肝脏和肾缺血再灌注损伤模型中,用MK4进行预处理都可改善器官损伤,减少脂质过氧化反应,降低细胞死亡。
3.维生素K可以转化成为氢醌 (VKH2),而氢醌是一种有效的自由基捕获抗氧化剂(RTA),可以抑制脂质过氧化。作者继续研究FSP1是否可以充当维生素K还原剂并产生VKH2以一直脂质过氧化,结果表明,FSP1可以介导维生素K还原,并维持VKH2作为RTA的功能。
4.作者进一步研究维生素K抑制铁死亡的机制,通过实验发现FSP1对维生素K的还原作用是叶绿醌和MK4抗铁死亡作用的原因。维生素K通过FSP1阻止铁死亡。
5.在血栓形成等疾病的治疗中通常warfarin作为抗凝剂,而warfarin也常常会带来无法凝血的副作用。使用高剂量维生素K是warfarin中毒的有效的解毒剂,由于FSP1可以还原维生素K,作者继续探究FSP1的酶活性是否可以介导warfarin耐受的维生素K还原途径。结果表明,FSP1是warfarin耐受的维生素 K 还原酶,可以防止warfarin毒性。
综上所述,这一系列研究表明维生素K可以通过其完全还原形式VKH2发挥抗铁死亡的作用,而FSP1是维生素还原酶,介导了维生素K抗铁死亡作用,和维生素K抗warfarin中度的作用,为维生素K在铁死亡及血栓形成相关疾病的治疗以及改善warfarin治疗中产生的副作用提供新的思路。
文章中使用到的技术主要有:细胞毒性分析,免疫组化,质谱分析,细胞凋亡,细胞迁移和侵袭
4、Cysteine depletion induces pancreatic tumor ferroptosis in mice
发表期刊:Science(IF:56.9)
发表时间:2020年4月
胰腺导管腺癌(PDAC)是一个致命的癌症并且对多种治疗效果耐受,超过90%的PDAC细胞会通过KRAS突变来促进自身的繁殖,这样的突变会增加脂质活性氧(ROS)的产生并且损伤细胞功能。为了自救,PDAC细胞通常会通过提高半胱氨酸衍生的代谢物如谷胱甘肽(GSH)来解除ROS毒性,避免自身死亡。来自哥伦比亚大学医疗中心的Kenneth P. Olive团队为我们揭示了半胱氨酸缺失可以诱导胰腺肿瘤铁死亡,抑制胰腺癌生长。
铁死亡是由ROS的灾难性积累导致,而半胱氨酸衍生的代谢物会抵消ROS的积累。作者发现,PDAC通过Glu/胱氨酸转运体(xC系统)输入氧化半胱氨酸(胱氨酸),同时利用半胱氨酸合成谷胱甘肽(glutathione)和辅酶A(coenzyme A)来抑制自身的铁死亡。缺失xC系统亚基Slc7a11或者通过胱氨酸酶类的药物都能够诱导PDAC细胞的铁死亡并抑制肿瘤生长。
文章的大概思路如下:
1.为了研究PDAC中的半胱氨酸代谢,作者在PDAC细胞中添加胱氨酸、xC系统的抑制剂(IKE)并测定细胞活力。实验结果发现半胱氨酸可增加细胞活力,xC系统的抑制剂使细胞存活率下降,同时铁死亡相关的抑制剂可减缓半胱氨酸耗竭而引起的细胞活力下降。并且半胱氨酸耗竭后细胞死亡前脂质氧化(铁死亡的一个标志)的大量增加,铁死亡相关的抑制剂同样也可以抵消,表明PDAC细胞中半胱氨酸耗竭将引发铁死亡。
2.作者用小鼠肿瘤模型进行进一步验证。结果表明在胰腺癌模型KPFSR小鼠中,xC系统的主要亚基Slc7a11基因敲除后肿瘤的生长减缓、小鼠的中位生存期增加了一倍。添加NAC(半胱氨酸类似物),小鼠的中位生存期又减少,并且肿瘤负荷增加。同时,Slc7a11的缺失,诱导了细胞的铁死亡,出现脂滴样球状上皮细胞和线粒体病变(铁死亡的标志)以及铁死亡相关基因的表达增加。
3.之前的研究表明,半胱氨酸主要通过GSH的合成来抑制细胞铁死亡,=然而单独BSO(GSH抑制剂)不能诱导PDAC细胞脂质氧化或降低细胞活力,这表明可能有其他半胱氨酸衍生代谢物有助于调控铁死亡。作者通过质谱检测标记的外源胱氨酸代谢物,发现胱氨酸在24小时内转化为辅酶A(CoA),而IKE可明显抑制CoA 水平,外源性CoA可以抑制IKE诱导的PDAC细胞铁死亡)。综上,联合抑制GSH和辅酶A(CoA)可诱导PDAC细胞铁死亡。
4.作者在细胞模型及肿瘤模型上验证半胱氨酸酶的作用。半胱氨酸酶诱导PDAC细胞系脂质氧化,降低细胞活力;其次,半胱氨酸酶治疗的小鼠的肿瘤组织明显出现铁死亡表型(脂滴形成、间质破坏),并且肿瘤体积相比对照组明显减小。
综上所述,作者通过一些列的研究表明某些癌症,例如PDAC依赖于半胱氨酸代谢抑制铁死亡,半胱氨酸缺失对于肿瘤生长有抑制作用,为半胱氨酸酶用于肿瘤治疗提供了新的研究思路。
文章中使用到的技术主要有:细胞毒性实验,流式细胞术,基因编辑,动物模型,液相色谱
5、Exosomes secreted from cardiomyocytes suppress the sensitivity of tumor ferroptosis in ischemic heart failure
发表期刊:Signal Transduction and Targeted Therapy(IF:39.3)
发表时间:2023年3月27日
心血管疾病和癌症一直以来都是严重危及生命的两大疾病,并且心力衰竭(HF)患者通常有较高的患癌风险。由于铁死亡在治疗耐药性癌症和其他退行性疾病方面发挥着重要作用,因此靶向铁死亡被认为是癌症治疗的新途径。外泌体对近端和远端器官通讯至关重要,并通过旁分泌方式调节疾病的发生。然而,外泌体是否能影响癌症对铁死亡的敏感性尚不清楚。来自哈尔滨医科大学的杨宝峰团队首次揭示了心肌梗死(MI)可以通过释放心肌细胞的miR-22-3p富集外泌体显著抑制ACSL4蛋白的表达,从而降低肿瘤细胞对铁死亡的敏感性。
心肌梗塞(MI)会降低癌细胞对铁死亡激活剂 erastin或 imidazole ketone erastin的敏感性。MI后血浆外泌体有效地减弱了肿瘤细胞对铁死亡诱导剂的敏感性。在慢性心肌梗死小鼠和高血压患者的衰竭心脏中,miR-22-3p 在心肌细胞和血浆外泌体中的表达明显上调。用从心肌梗死后小鼠血浆中分离出的外泌体培养肿瘤细胞或单独过表达miR-22-3p可减轻erastin诱导的铁死亡。心肌细胞的miR-22-3p会被外泌体包装并转移到肿瘤细胞中。用 AAV9 sponge抑制心肌细胞特异性 miR-22-3p增加了癌细胞对铁死亡的敏感性。促铁死亡基因 ACSL4 是 miR-22-3p在肿瘤细胞中的靶点。
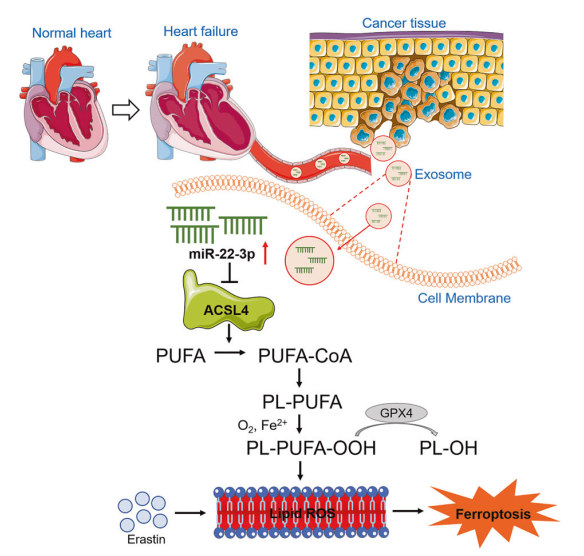
文章的大概思路如下:
1.为了阐明心肌梗死后(post-MI)的心力衰竭(HF)对癌细胞铁死亡敏感性的影响,作者使用铁死亡诱导剂erastin和IKE,在sham或MI实验模型中抑制移植瘤的生长。结果表明,MI在很大程度上抑制了erastin或IKE诱导的小鼠肿瘤生长的减少。MI诱导的HF在体内抑制了肿瘤对铁死亡的敏感性。
2.作者继续研究MI影响癌细胞对铁死亡敏感性的具体机制。研究表明,外泌体会影响癌症的进程,作者决定研究HF中的外泌体是否会影响erastin诱导的铁死亡,结果表明,erastin在LLC移植瘤模型中会诱导脂质过氧化并抑制肿瘤生长,而用MI后的外泌体(MI-EXO)处理可显著恢复。MI外泌体也可以逆转erastin引起的Ki67降低和4-HNE增加。结果表明,来自MI诱导的HF的外泌体可以降低肿瘤对铁死亡的敏感性。
3.作者继续在体外用细胞系进行验证。作者分离了Sham-EXOS和MI-EXOS并与小鼠LLC肺癌细胞系或K7M2骨肉瘤细胞系共培养,结果表明,erastin明显增加了脂质ROS积累,而MI-EXO减轻了这一作用和erastin诱导的铁死亡,MI-EXO处理的细胞内铁离子水平普遍下降。此外,MI-EXO抑制铁死亡,促进癌细胞增殖、侵袭和迁移。以上结果表明外泌体可抑制癌症细胞铁死亡。
4.为了更好地了解外泌体的病理机制,作者分析了缺血心肌中miRNA的含量,发现miR-22-3p含量最丰富,此外,小鼠缺血心肌和血浆外泌体中的miR-22-3p水平均上调。这些结果表明,MI-EXO的miR-22-3p减弱了癌细胞对erastin诱导的铁死亡的敏感性。
5.为了证实功能性血浆外泌体主要来源于MI诱导的衰竭心肌细胞,作者从Sham-Myo和MI-Myo小鼠的成年心室肌细胞中获取并纯化了心肌外泌体Myo-EXOSham和Myo-EXOMI,发现MI-EXO/Myo-EXOMI的促癌作用确实是通过miR-22-3p介导的,并且内源性miR-223p也抑制了肿瘤细胞的铁死亡。
6.接下来,作者对MiR-22-3p相关的靶基因进行验证,作者筛选出了MiR-22-3p作用的靶基因acyl-CoA synthetase long-chain family member 4(ACSL4)以及结合位点,并通过实验验证了miR-22-3p通过靶向ACSL4抑制erastin诱导的肿瘤细胞铁死亡。
综上所述,MI-EXO在降低癌症细胞对铁死亡的敏感性中起到了重要的作用并且会促进肿瘤的生长,外泌体的miR-22-3p下调ACSL44是抑制癌症细胞对铁死亡敏感性的分子机制,这一系列研究也为靶向外泌体介导的心肌细胞/肿瘤病理通讯基于铁死亡的抗肿瘤治疗提供了新的思路。
文章中使用到的技术主要有:超声心动图系统,移植瘤模型,外泌体分离,纳米粒子跟踪分析(NTA),透射电子显微镜,迁移和侵袭试验



