
 |
|
|||||||||||||||||||||||||||||
| [发表评论] [本类其他产品] [本类其他供应商] [收藏] | ||||||||||||||||||||||||||||||
| 销售商: 北京百奥知信息科技有限公司 | 查看该公司所有产品 >> |
一、产品概述
您在生物数据分析是否遇到过下面令人头疼的问题呢?
•现在的生物、医学、药学等研究都会遇到数据处理,数据如何分析才能得到好的结果…
•高通量技术在生物各个学科应用中越来普遍,如何进行实验设计…
•实验得到的芯片数据、测序数据、质谱数据如何分析…
•自己的实验数据分析流程是什么…
•同一个处理过程,分析方法太多,哪个结果更好…
•分析结果如何解释生物学功能…
百奥知数据分析系统(Bioknow Data Analysis System,简称Bioknow-DataAnalysis),是生物数据分析软件系统。系统提供各种生物数据平台的数据分析支持,如表达谱芯片、miRNA芯片、SNP芯片、比较基因组杂交芯片、蛋白芯片、Promoter芯片以及Affymetrix提供的各种芯片等,并提供各种常用的归一化方法、检验方法、聚类方法、分类方法、调控网络重构方法、各种相关图形绘制等;同时系统还支持新测序数据如RNA-seq、Chiq-seq、miRNA-seq等进行序列拼接、序列定位、表达水平评估、差异表达分析等;此外系统还提供医学常用的统计分析,例如假设检验、方差分析、卡方检验、回归分析、PCA分析、生存分析等。旨在为医药研究领域分析人员以及生物信息学研究者提供各种数据分析的平台,发掘基因表达与其功能间的关系,帮助研究者发现新药靶点、开展疾病的基因诊断和早期预警等。
二、应用范围
大规模实验数据的生物学分析;
各类生物分子的关联功能统计分析;
三、产品功能

图 产品功能图
3.1 生物数据分析
实现表达谱芯片数据的预处理、归一化、荧光交换数据分析等
实现多种常用的统计学检验方法和聚类/分类方法,实现差异基因的挑选
实现多种类型芯片数据的整合分析方法,供试验者从不同角度数据进行综合分析
实现多种类型芯片数据的整合分析方法,供试验者从不同角度数据进行综合分析
芯片数据概要模块
M vs. A:芯片数据分布
Histogram:单张芯片的数据分布情况
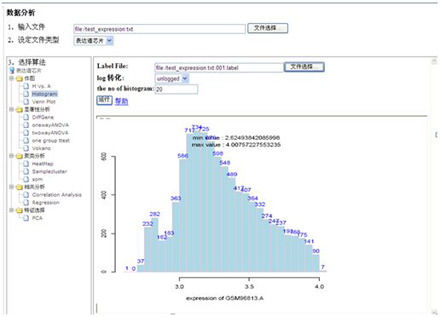
图 芯片数据分布直方图
显著性分析模块
DiffGene:组合t-test、SAM分析
One group ttest:单组样本T检验
onewayANOVA:单因素方差分析
twowayANOVA:双因素方差分析
Vokano:显著型结果作图
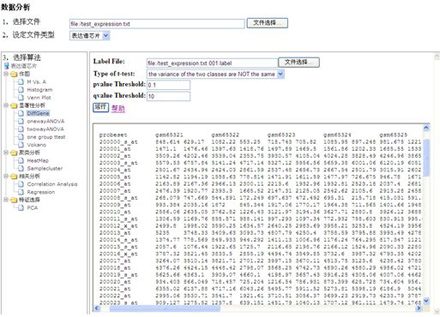
图 差异表达基因分析
多基因集分析模块包括:
Venn Plot:计算三个基因之间的交集,并集

图 多基因集关系分析
聚类分析的模块:
HeatMap:对样本和基因进行双向聚类
Sample cluster:对样本进行层次聚类
SOM:自组织映射聚类
其他分析模块:
Compare:计算样本间的相似度
PCA:主成份分析
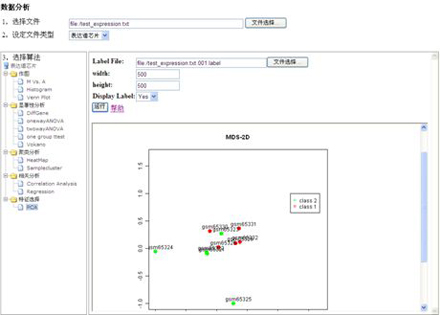
图 PCA分析结果
3.2 疾病相关SNP分析
数据基本情况,检验Hardy-Weinberg平衡,MAF和位点非缺失基因型所占百分比等位点相关信息。
SNP关联分析,通过 Pearson 卡方检验或Fisher精确检验,分析正常组和疾病组每个位点上基因型和等位基因的分布差异,寻找与疾病相关的位点。
LD(连锁不平衡)分析,这种不同基因座位的某些等位基因非随机联合经常会在一起遗传。通过D’/r^2等考察位点之间的连锁不平衡。
单体型分析,通过Pearson 卡方检验考察这些整体遗传的单体型是否和疾病关联(p值<0.05)。
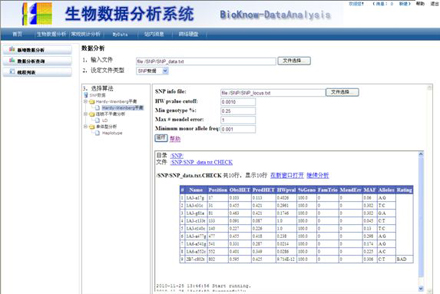
图 HW平衡检验
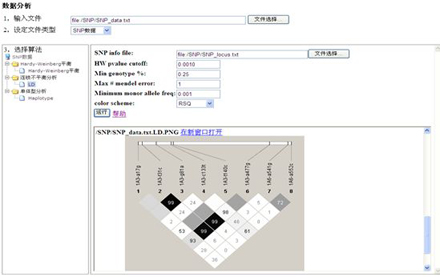
图 连锁不平衡分析
3.3 常见统计分析
医学统计是运用统计学的原理和方法研究医学科研中有关数据的收集、整理和分析。正确运用统计学思维,针对数据特点,巧妙地选用恰当高效的统计分析方法,从而得到可靠的结果和科学的结论。
统计分析模块
假设检验:T检验
方差分析:ANOVA
非参数检验:sampleks、kappa、chisquare、pairchisq
相关分析:correlation、distance、linear regression
聚类分析:kmeans、 hierarchy、 SOM
主成份分析:PCA
生存分析:survival
3.4 高通亮测序数据分析
Solexa数据处理
Solexa数据处理包括图片分析、碱基识别、序列分析。通过数据处理,测序仪上产生的图片文件被转换为易于进行下一步分析的序列文件和质量文件;维护与改进标准数据处理流程;对于可能影响数据质量的所有因素,寻求解决方案,并对数据处理与质控提供技术支持。
RNA-Seq测序分析方案
基因组定位,数据处理由原始fastq文件开始。数据首先将含有Q<20的碱基替换为N。然后使用MAQ软件将fastq转换为Fasta。然后去掉3’-adaptor。然后序列数据通过MAQ软件将序列向基因组比对。生成覆盖度数据。
转录本定量分析,MAQ比对的结果用于基因定量,对于每个基因进行基因定量,定量方法采用 reads per kilobase of exon per million mapped sequence reads (RPKM)反映表达量。
可变剪切预测,我们使用TopHat软件进行进行可变剪切的分析。
反义转录本的寻找。
识别新转录本、选择性剪切和组织特异性microRNA信息,通过将Reads往参考基因组上Mapping,可以将Reads定位到考基因组三类位置:已经被标注的基因外显子区域;已经被发现的非编码RNA区域;未被确认的区域。
获取SNP、Indel和Mutation数据,统计不同个体、组织间的分布差异,方法为基于SNP、Indel和Mutation位点reads覆盖度的统计检验方法筛选在组织间具有显著差异的位点或者基因。
sRNA测序分析方案
将测序结果分为microRNA, piRNA, tRNA, snRNA, rRNA, snoRNA 等,最后绘制成饼图展示结果。
计算已知miRNA的表达量,对于处理-对照实验设计采用Audic Claverie test筛选两个样本间的差异表达microRNA。
miRNA家族、基因座分类,有一些MicroRNA在基因组中成簇(cluster)分布。这些microRNA同步转录。由于转录后成熟过程的调节各异,因此成簇的microRNA成熟体的表达量略有不同。通过对成簇microRNA的分析,可以在cluster的水平考察microRNA的差异表达情况。
预测新的miRNA, 根据solexa测序得到的未知序列信息,结合基因组提供的序列上下文信息,使用miRDeep预测solexa测序得到的未知序列中可能存在的novel microRNA。
miRNA进化分析,跨物种比较microRNA 序列的保守性及种子序列的保守性。
microRNA 基因簇分析,将新预测的microRNA 在基因组上定位,并寻找可能同时转录的microRNA。
mRNA测序分析方案
基因注释,我们将测序结果进行比对和分析,确定tag代表的基因。
差异基因筛选,筛选到表达有差异变化的基因。然后对变化基因的趋势进行归类,以方便分析和描述。
样品之间的比较分析,我们将通过曲线拟合的方法,寻找样品之间趋势差异最大的一些基因。
GeneOntology分析,对于每一种表达趋势的基因,选择性的进gene ontology:功能分析. 对差异表达的所有基因向gene ontology数据库的各节点映射。计算每个节点的基因数目,并结合整个数据库的基因作为背景分部,对于每个节点,得到一个2x2的表格,使用超几何分布检验基因在每个GO节点的富集或贫乏程度。
Pathway分析,我们同样将差异基因使用KEGG pathway和BioCarta数据库映射。通过统计方案,找到统计上最有意义的pathway。
转录因子分析,利用相关的转录因子(TF)数据库,将差异基因的启动子(基因第一个外显子上游1000bp)提取出来。我们使用HsPD 提供的启动子序列。转录因子数据库使用TRANSFAC 7.0 public。转录因子结合位点预测使用pwmatch 程序,对每个转录因子分析其在上调基因和下调基因的分布情况,利用chi-square test 寻找有差异的转录因子。
ChIP-Seq单通道(Lane)Single-end数据信息分析方案
将测序结果进行大规模随机blast,检测可能的样品污染。
Reads比对到基因组,将测序结果与reference genome进行mapping, mapping 的reads 中mismatch 的数目可以根据客户需要进行分析, 并挑选出unique mapping 的所有reads用于后续peak的分析。
Peak的查找,根据mapping的unique reads进行peak查找, 确定具有统计学意义的靶位点,用于后续分析。
Peak的全基因组定位,对查找的具有统计学意义的peak进行全基因组locus定位,在得到每个peak对应的locus的基础上,再对全基因组上peak进行位置的分布统计,以对靶基因的偏好性进行了解。
Peak附近区域序列提取及motif分析,对peak附近的区域进行序列提取,并对其做motif分析,特别是对转录因子结合位点测序,在验证已有motif的基础上,进行denovo分析,试图发现新的motif。
靶基因的GO,PATHWAY,NETWORK分析。
